

The role of the NLRP3 inflammasome and pyroptosis in cardiovascular diseases
- PMID: 37923829
- PMCID: PMC11550901
- DOI: 10.1038/s41569-023-00946-3
The role of the NLRP3 inflammasome and pyroptosis in cardiovascular diseases
Abstract
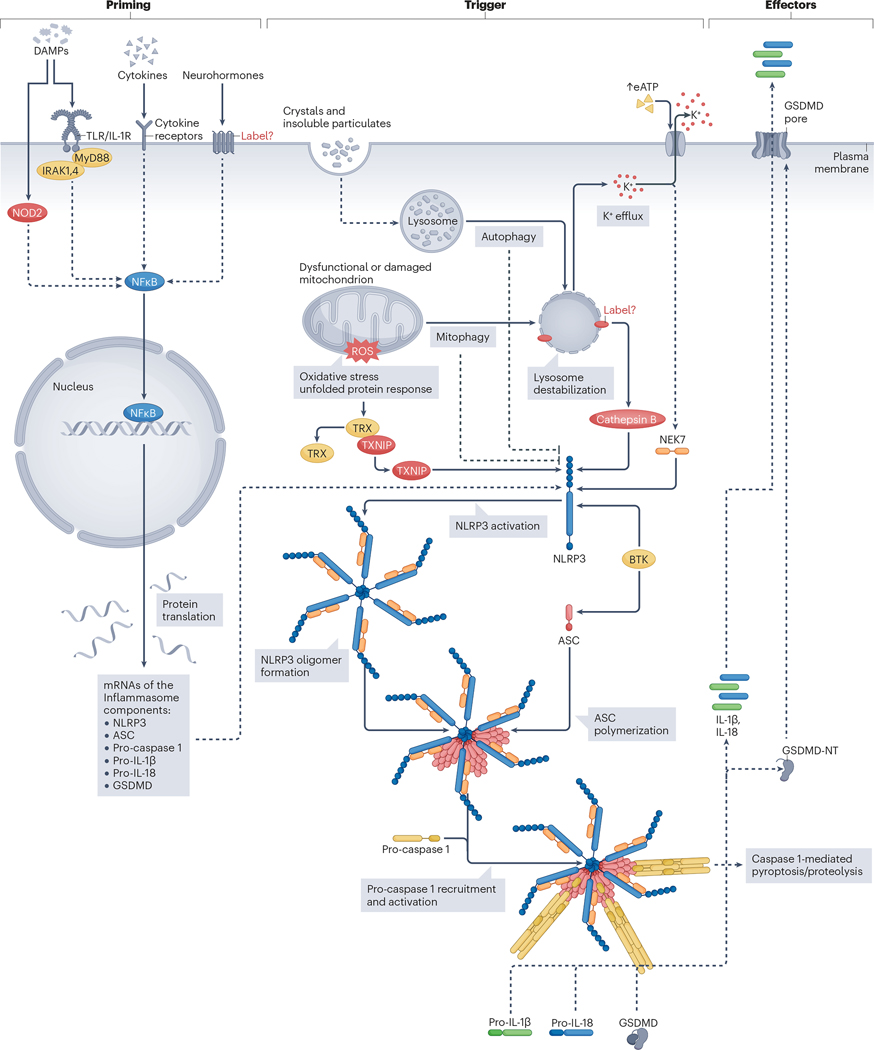
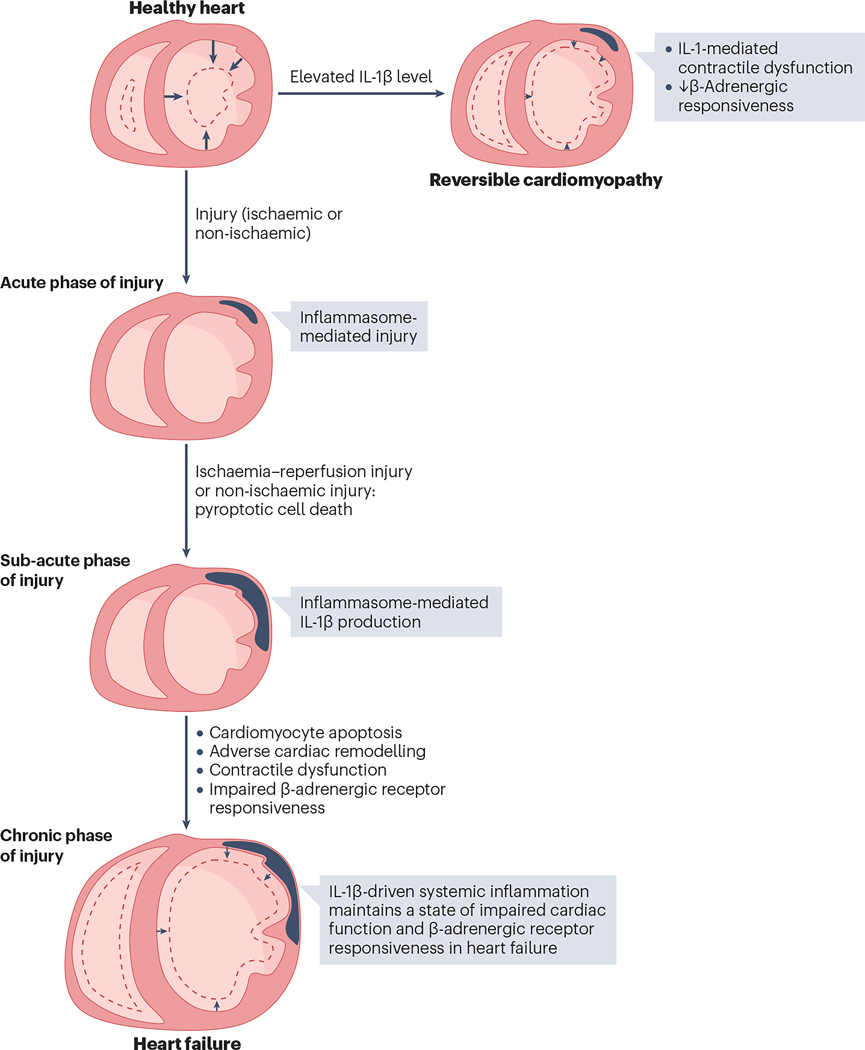
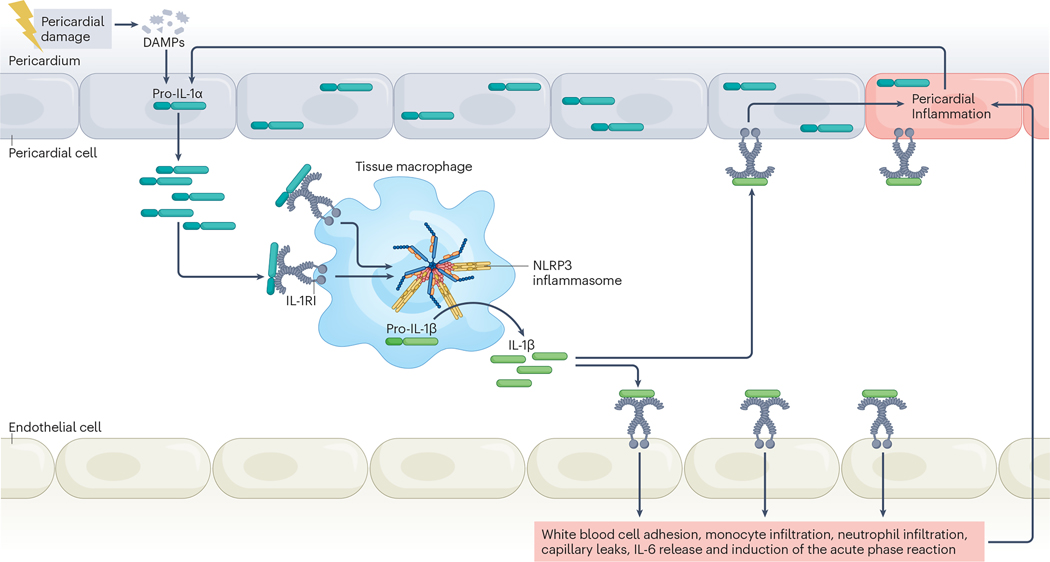
An intense, stereotyped inflammatory response occurs in response to ischaemic and non-ischaemic injury to the myocardium. The NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome is a finely regulated macromolecular protein complex that senses the injury and triggers and amplifies the inflammatory response by activation of caspase 1; cleavage of pro-inflammatory cytokines, such as pro-IL-1β and pro-IL-18, to their mature forms; and induction of inflammatory cell death (pyroptosis). Inhibitors of the NLRP3 inflammasome and blockers of IL-1β and IL-18 activity have been shown to reduce injury to the myocardium and pericardium, favour resolution of the inflammation and preserve cardiac function. In this Review, we discuss the components of the NLRP3 inflammasome and how it is formed and activated in various ischaemic and non-ischaemic cardiac pathologies (acute myocardial infarction, cardiac dysfunction and remodelling, atherothrombosis, myocarditis and pericarditis, cardiotoxicity and cardiac sarcoidosis). We also summarize current preclinical and clinical evidence from studies of agents that target the NLRP3 inflammasome and related cytokines.
© 2023. Springer Nature Limited.
Conflict of interest statement
Competing interests
A.A. has served as a consultant to Cardiol, Implicit Bioscience, Janssen, Kiniksa, Novo Nordisk, Olatec, R-Pharm, Sanofi and Serpin Pharma. S.T. has received research grants from Cardiol, Kiniksa and Olatec.
Figures




References
-
- Tsao CW et al. Heart Disease and Stroke Statistics-2022 Update: A Report From the American Heart Association. Circulation 145, e153–e639 (2022). - PubMed
-
- Lenz A, Franklin GA & Cheadle WG Systemic inflammation after trauma. Injury 38, 1336–1345 (2007). - PubMed
-
- Toldo S. & Abbate A. The NLRP3 inflammasome in acute myocardial infarction. Nat. Rev. Cardiol 15, 203–214 (2018). - PubMed
-
- Westman PC et al. Inflammation as a Driver of Adverse Left Ventricular Remodeling After Acute Myocardial Infarction. J. Am. Coll. Cardiol 67, 2050–2060 (2016). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
