

Efficient in vivo prime editing corrects the most frequent phenylketonuria variant, associated with high unmet medical need
- PMID: 37924808
- PMCID: PMC10716342
- DOI: 10.1016/j.ajhg.2023.10.005
Efficient in vivo prime editing corrects the most frequent phenylketonuria variant, associated with high unmet medical need
Abstract
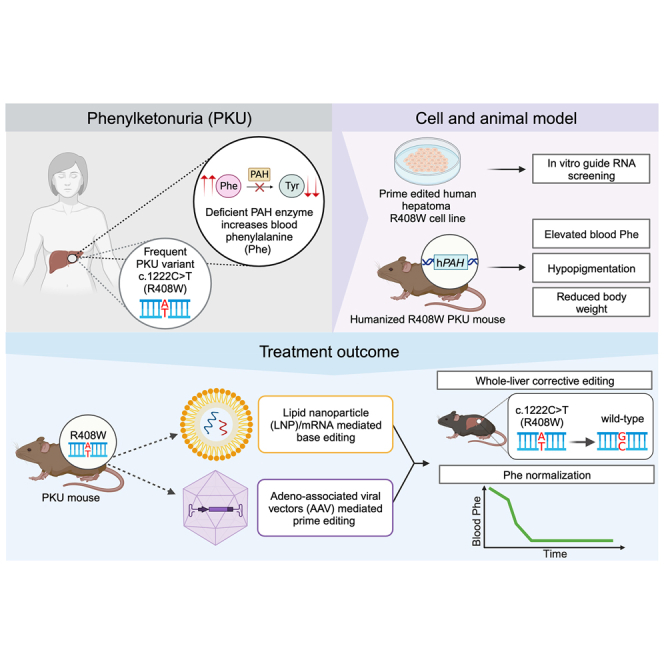
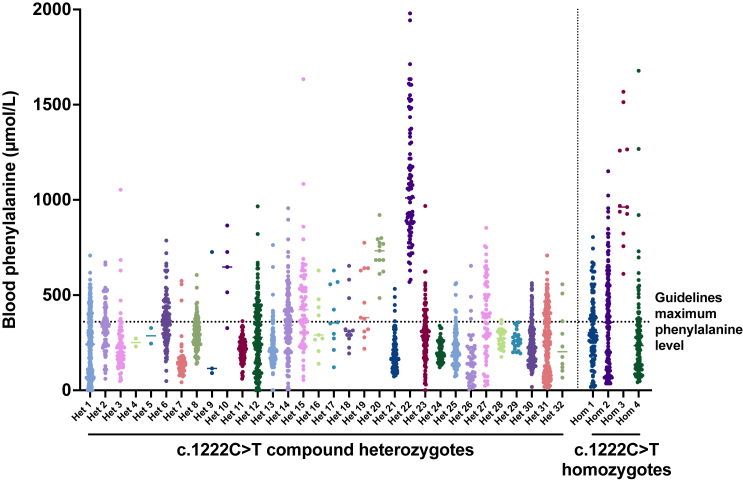
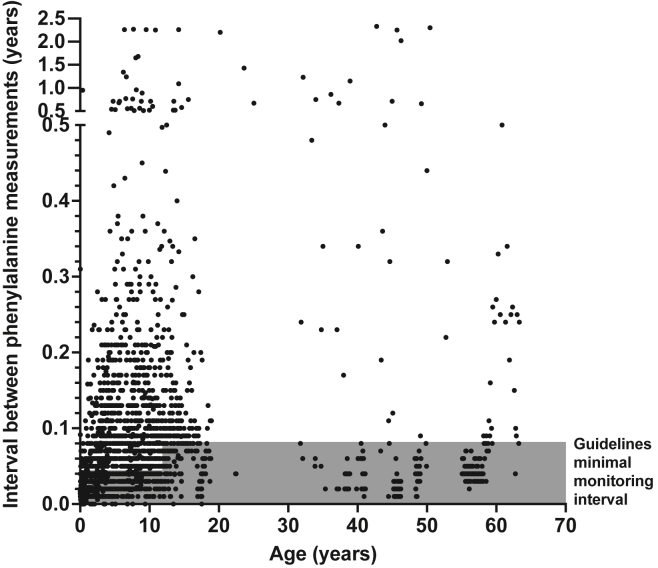
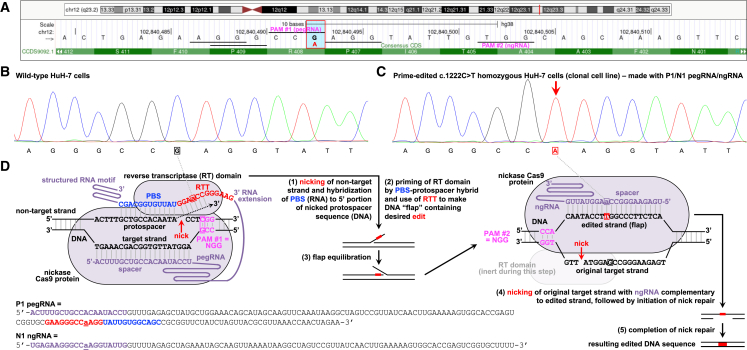
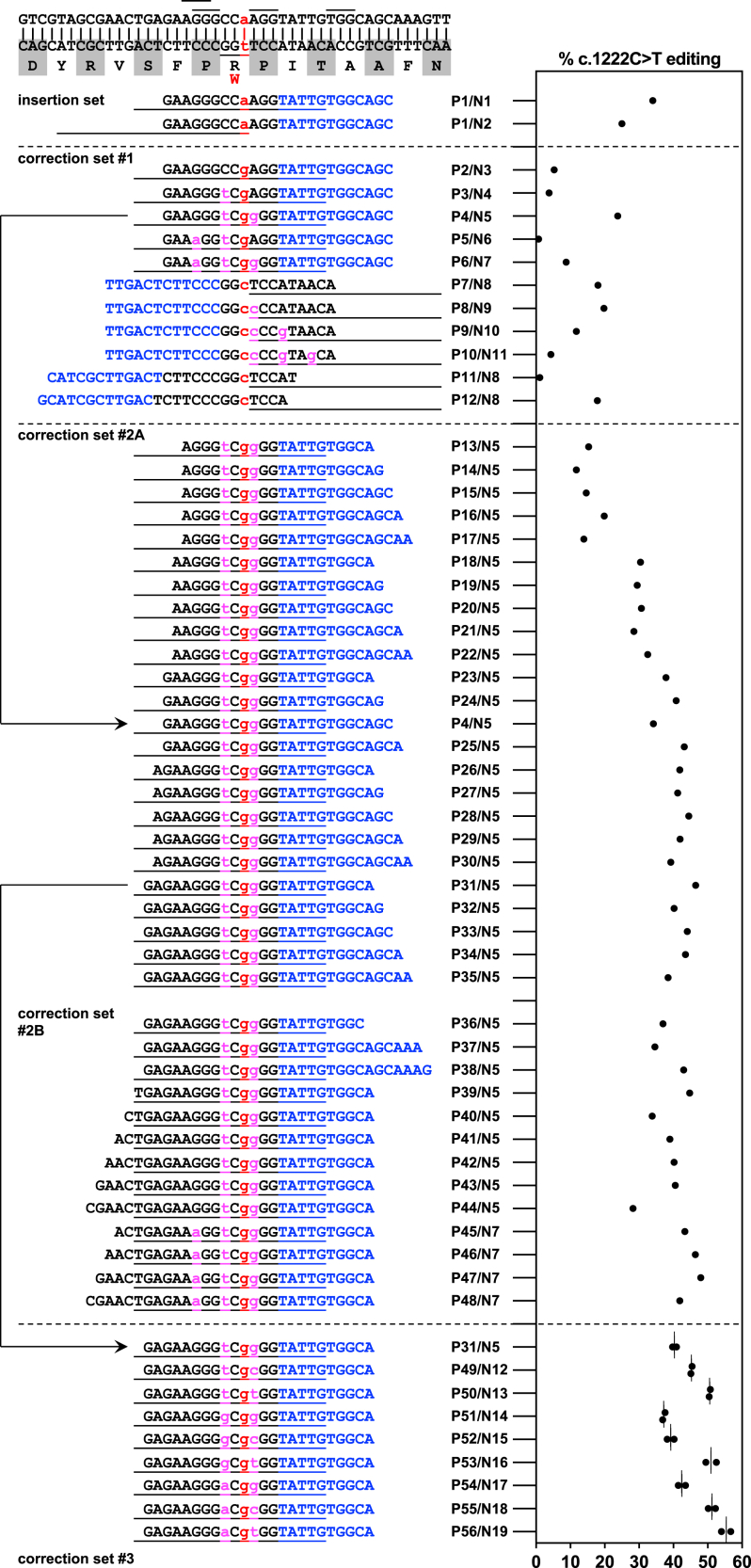
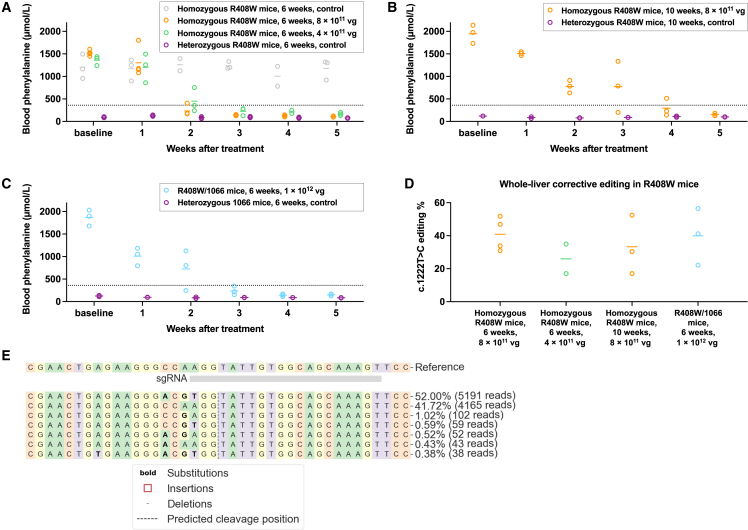
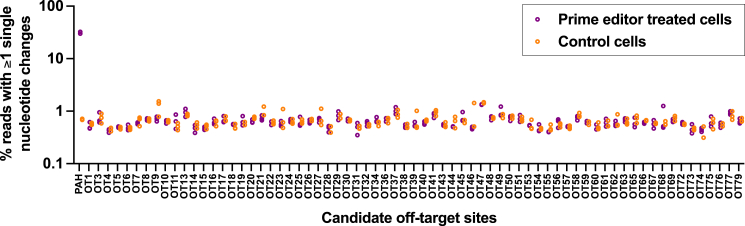
The c.1222C>T (p.Arg408Trp) variant in the phenylalanine hydroxylase gene (PAH) is the most frequent cause of phenylketonuria (PKU), the most common inborn error of metabolism. This autosomal-recessive disorder is characterized by accumulation of blood phenylalanine (Phe) to neurotoxic levels. Using real-world data, we observed that despite dietary and medical interventions, most PKU individuals harboring at least one c.1222C>T variant experience chronic, severe Phe elevations and do not comply with Phe monitoring guidelines. Motivated by these findings, we generated an edited c.1222C>T hepatocyte cell line and humanized c.1222C>T mouse models, with which we demonstrated efficient in vitro and in vivo correction of the variant with prime editing. Delivery via adeno-associated viral (AAV) vectors reproducibly achieved complete normalization of blood Phe levels in PKU mice, with up to 52% whole-liver corrective PAH editing. These studies validate a strategy involving prime editing as a potential treatment for a large proportion of individuals with PKU.
Keywords: CRISPR; gene editing; genome editing; inborn error of metabolism; metabolic disease; phenylketonuria; prime editing; rare disease.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests K.M. is an advisor to and holds equity in Verve Therapeutics and Variant Bio and is an advisor to LEXEO Therapeutics. R.C.A.-N. is an advisor to Latus Bio. All other authors have no financial conflicts of interests to disclose.
Figures
Comment in
-
A base editing strategy using mRNA-LNPs for in vivo correction of the most frequent phenylketonuria variant.HGG Adv. 2024 Jan 11;5(1):100253. doi: 10.1016/j.xhgg.2023.100253. Epub 2023 Nov 2. HGG Adv. 2024. PMID: 37922902 Free PMC article.
References
-
- Vockley J., Andersson H.C., Antshel K.M., Braverman N.E., Burton B.K., Frazier D.M., Mitchell J., Smith W.E., Thompson B.H., Berry S.A., American College of Medical Genetics and Genomics Therapeutics Committee Phenylalanine hydroxylase deficiency: diagnosis and management guideline. Genet. Med. 2014;16:188–200. doi: 10.1038/gim.2013.157. - DOI - PubMed
-
- Jurecki E.R., Cederbaum S., Kopesky J., Perry K., Rohr F., Sanchez-Valle A., Viau K.S., Sheinin M.Y., Cohen-Pfeffer J.L. Adherence to clinic recommendations among patients with phenylketonuria in the United States. Mol. Genet. Metabol. 2017;120:190–197. doi: 10.1016/j.ymgme.2017.01.001. - DOI - PubMed
-
- Leuders S., Wolfgart E., Ott T., du Moulin M., van Teeffelen-Heithoff A., Vogelpohl L., Och U., Marquardt T., Weglage J., Feldmann R., Rutsch F. Influence of PAH genotype on sapropterin response in PKU: results of a single-center cohort study. JIMD Rep. 2014;13:101–109. doi: 10.1007/8904_2013_263. - DOI - PMC - PubMed
-
- Burton B.K., Longo N., Vockley J., Grange D.K., Harding C.O., Decker C., Li M., Lau K., Rosen O., Larimore K., et al. Pegvaliase for the treatment of phenylketonuria: results of the phase 2 dose-finding studies with long-term follow-up. Mol. Genet. Metabol. 2020;130:239–246. doi: 10.1016/j.ymgme.2020.06.006. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
