

Spatial transcriptomic interrogation of the murine bone marrow signaling landscape
- PMID: 37926705
- PMCID: PMC10625929
- DOI: 10.1038/s41413-023-00298-1
Spatial transcriptomic interrogation of the murine bone marrow signaling landscape
Abstract
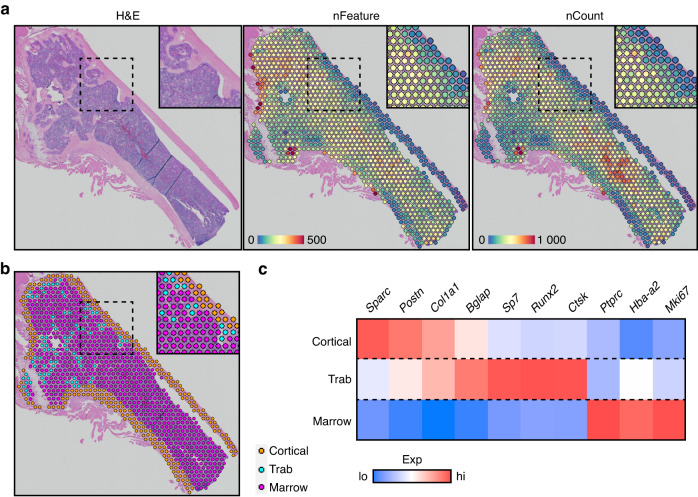
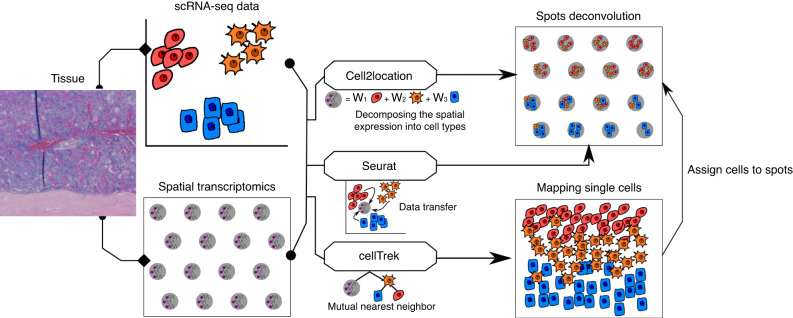
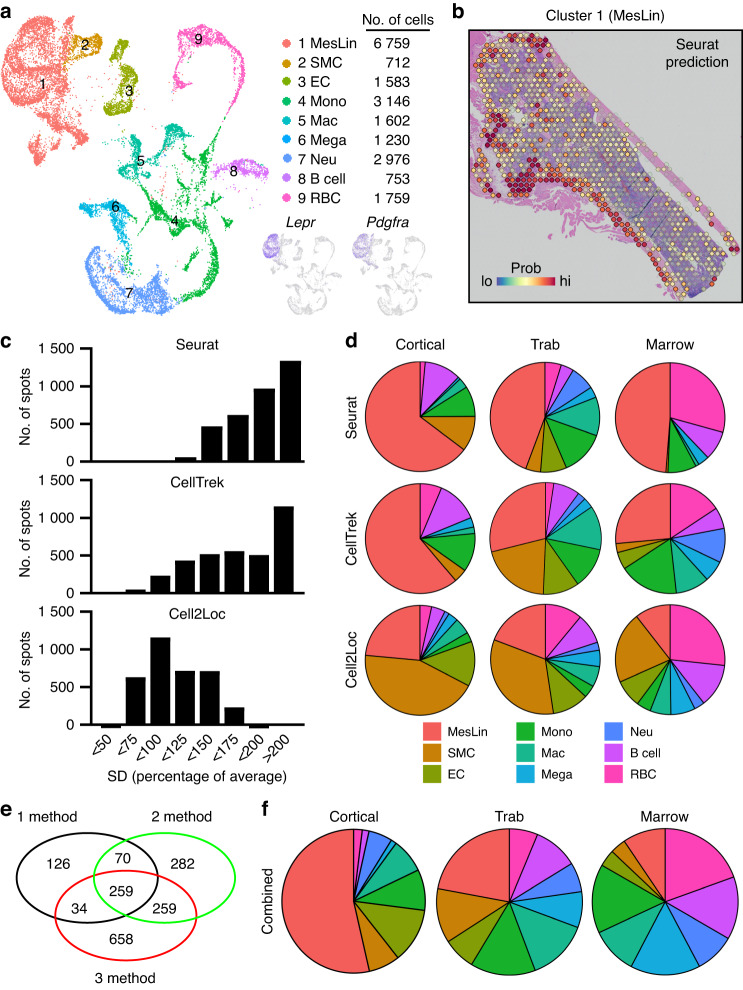
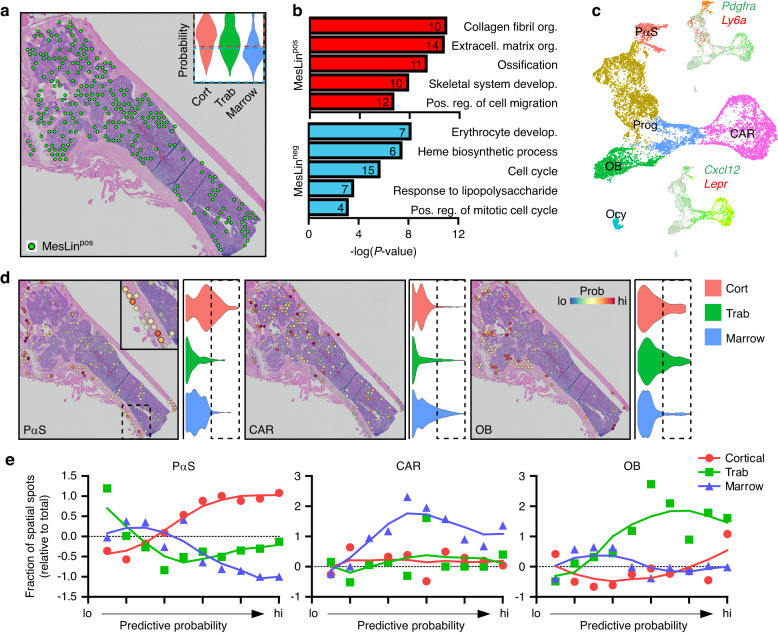
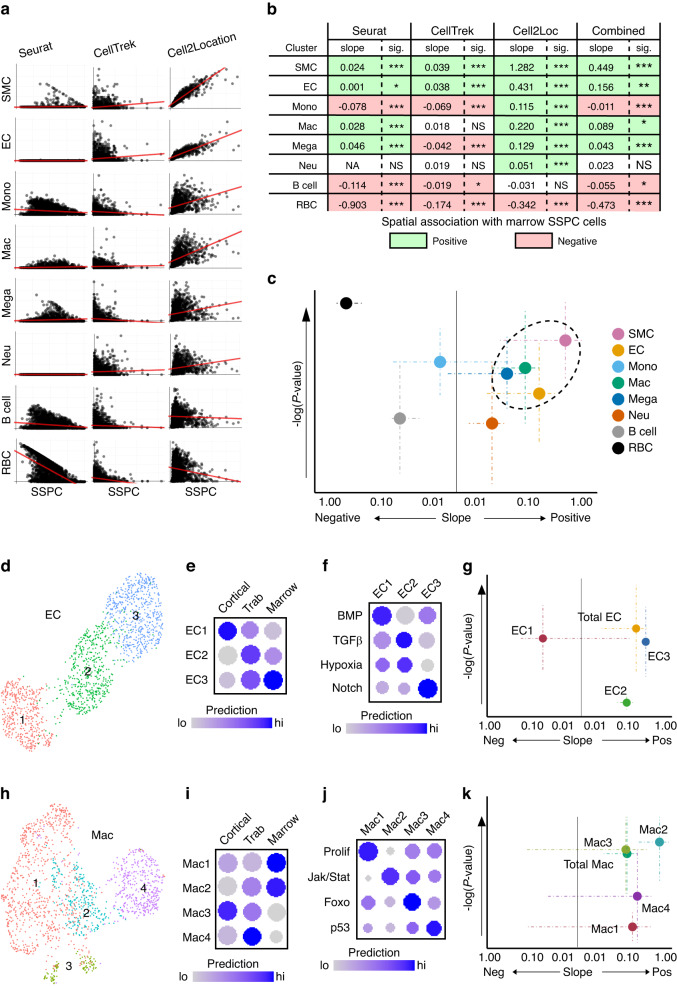
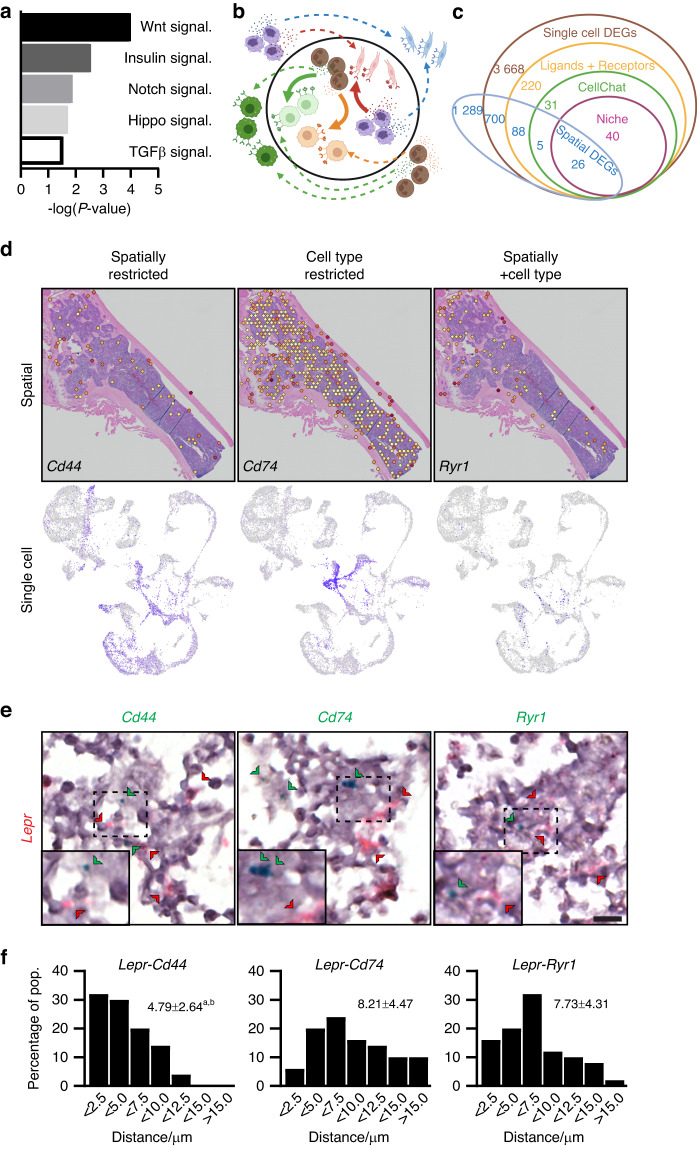
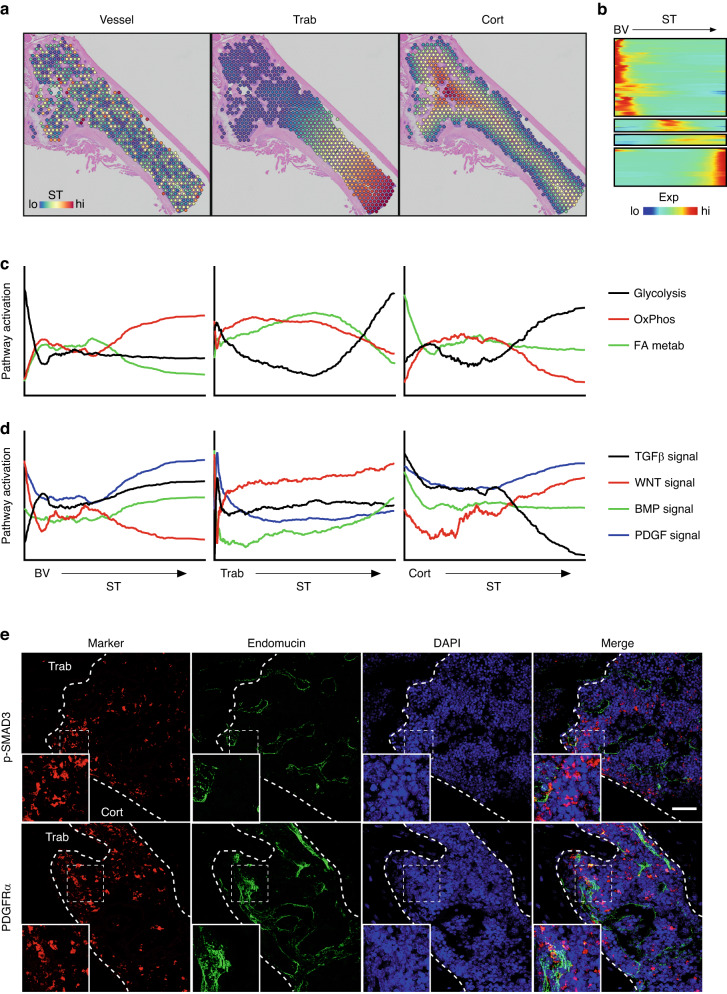
Self-renewal and differentiation of skeletal stem and progenitor cells (SSPCs) are tightly regulated processes, with SSPC dysregulation leading to progressive bone disease. While the application of single-cell RNA sequencing (scRNAseq) to the bone field has led to major advancements in our understanding of SSPC heterogeneity, stem cells are tightly regulated by their neighboring cells which comprise the bone marrow niche. However, unbiased interrogation of these cells at the transcriptional level within their native niche environment has been challenging. Here, we combined spatial transcriptomics and scRNAseq using a predictive modeling pipeline derived from multiple deconvolution packages in adult mouse femurs to provide an endogenous, in vivo context of SSPCs within the niche. This combined approach localized SSPC subtypes to specific regions of the bone and identified cellular components and signaling networks utilized within the niche. Furthermore, the use of spatial transcriptomics allowed us to identify spatially restricted activation of metabolic and major morphogenetic signaling gradients derived from the vasculature and bone surfaces that establish microdomains within the marrow cavity. Overall, we demonstrate, for the first time, the feasibility of applying spatial transcriptomics to fully mineralized tissue and present a combined spatial and single-cell transcriptomic approach to define the cellular components of the stem cell niche, identify cell‒cell communication, and ultimately gain a comprehensive understanding of local and global SSPC regulatory networks within calcified tissue.
© 2023. The Author(s).
Conflict of interest statement
Spatial data were generated in cooperation with 10x Genomics, the manufacturer of the Visium Spatial Gene Expression system.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
