

Identification and functional characterization of de novo variant in the SYNGAP1 gene causing intellectual disability
- PMID: 37928246
- PMCID: PMC10622656
- DOI: 10.3389/fgene.2023.1270175
Identification and functional characterization of de novo variant in the SYNGAP1 gene causing intellectual disability
Abstract
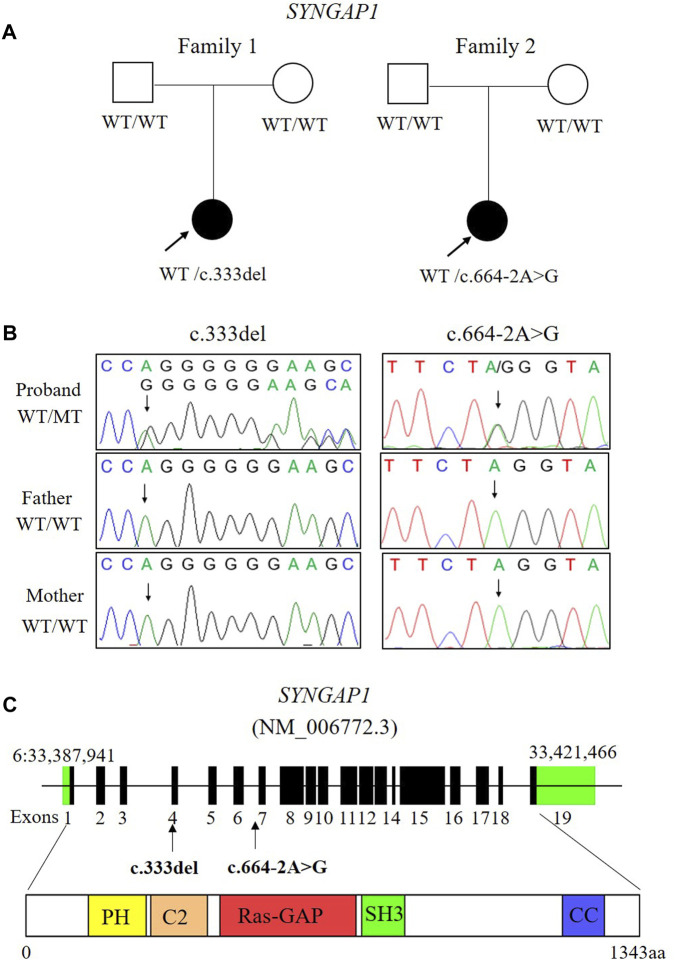
Background: Intellectual disability (ID) is defined by cognitive and social adaptation defects. Variants in the SYNGAP1 gene, which encodes the brain-specific cytoplasmic protein SYNGAP1, are commonly associated with ID. The aim of this study was to identify novel SYNGAP1 gene variants in Chinese individuals with ID and evaluate the pathogenicity of the detected variants. Methods: Whole exome sequencing (WES) was performed on 113 patients diagnosed with ID. In the study, two de novo variants in SYNGAP1 were identified. Sanger sequencing was used to confirm these variants. Minigene assays were used to verify whether the de novo intronic variant in SYNGAP1 influenced the normal splicing of mRNA. Results: Two de novo heterozygous pathogenic variants in SYNGAP1, c.333del and c.664-2A>G, were identified in two ID patients separately. The c.333del variant has been reported previously as a de novo finding in a child with ID, while the c.664-2A>G variant was novel de novo intronic variant, which has not been reported in the literature. Functional studies showed that c.664-2A>G could cause aberrant splicing, resulting in exon 7 skipping and a 16bp deletion within exon 7. Conclusion: We identified two de novo pathogenic heterozygous variants in SYNGAP1 in two patients with ID, among which the c.664-2A>G variant was a novel de novo pathogenic variant. Our findings further enrich the variant spectrum of the SYNGAP1 gene and provide a research basis for the genetic diagnosis of ID.
Keywords: SYNGAP1; intellectual disability; minigene; variant; whole exome sequencing (WES).
Copyright © 2023 Li, Wang, Hou, Song, Zhang, Li, Yang and Sun.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures


Similar articles
-
Novel De Novo Intronic Variant of SYNGAP1 Associated With the Neurodevelopmental Disorders.Mol Genet Genomic Med. 2025 Feb;13(2):e70066. doi: 10.1002/mgg3.70066. Mol Genet Genomic Med. 2025. PMID: 39878419 Free PMC article.
-
Molecular and phenotypical findings of a novel de novo SYNGAP1 gene variant in an 11-year-old Iranian boy with intellectual disability.Lab Med. 2024 Mar 7;55(2):204-208. doi: 10.1093/labmed/lmad064. Lab Med. 2024. PMID: 37467311
-
Novel variants in the CLCN4 gene associated with syndromic X-linked intellectual disability.Front Neurol. 2023 Sep 15;14:1096969. doi: 10.3389/fneur.2023.1096969. eCollection 2023. Front Neurol. 2023. PMID: 37789889 Free PMC article.
-
Trio exome sequencing identified a novel de novo WASF1 missense variant leading to recurrent site substitution in a Chinese patient with developmental delay, microcephaly, and early-onset seizures: A mutational hotspot p.Trp161 and literature review.Clin Chim Acta. 2021 Dec;523:10-18. doi: 10.1016/j.cca.2021.08.030. Epub 2021 Aug 31. Clin Chim Acta. 2021. PMID: 34478686 Review.
-
Novel SYNGAP1 variant in a patient with intellectual disability and distinctive dysmorphisms.Congenit Anom (Kyoto). 2018 Nov;58(6):188-190. doi: 10.1111/cga.12273. Epub 2018 Feb 13. Congenit Anom (Kyoto). 2018. PMID: 29381230 Review.
References
LinkOut - more resources
Full Text Sources