

Stem cell-like reprogramming is required for leukemia-initiating activity in B-ALL
- PMID: 37930337
- PMCID: PMC10626194
- DOI: 10.1084/jem.20230279
Stem cell-like reprogramming is required for leukemia-initiating activity in B-ALL
Abstract
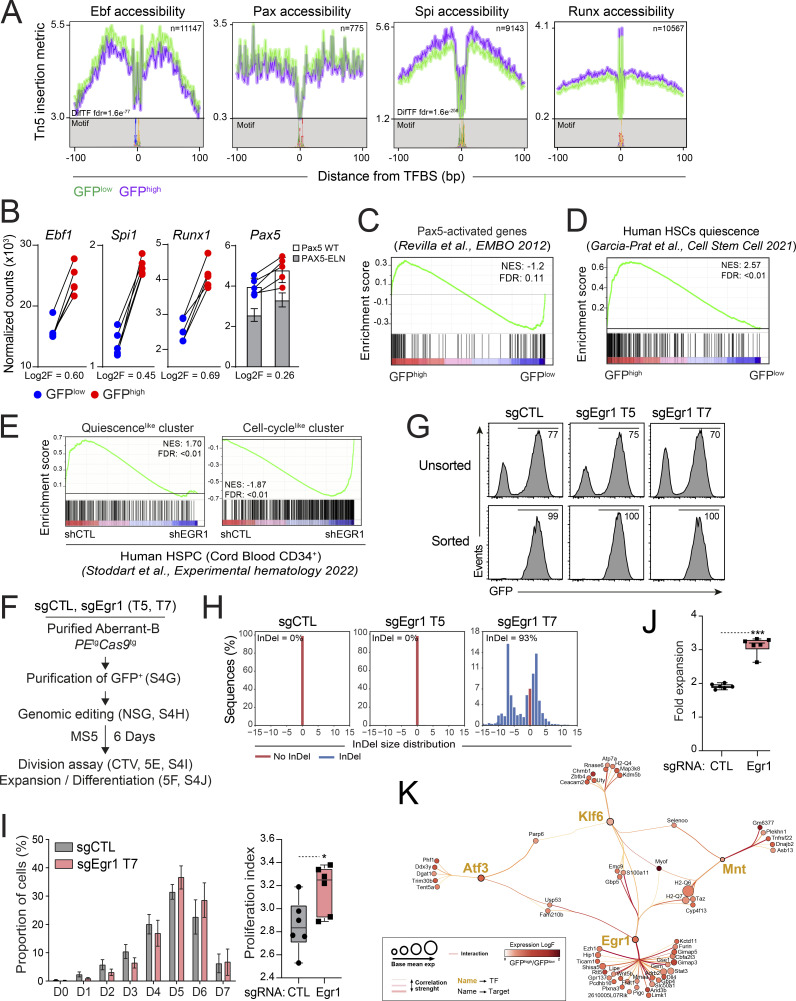
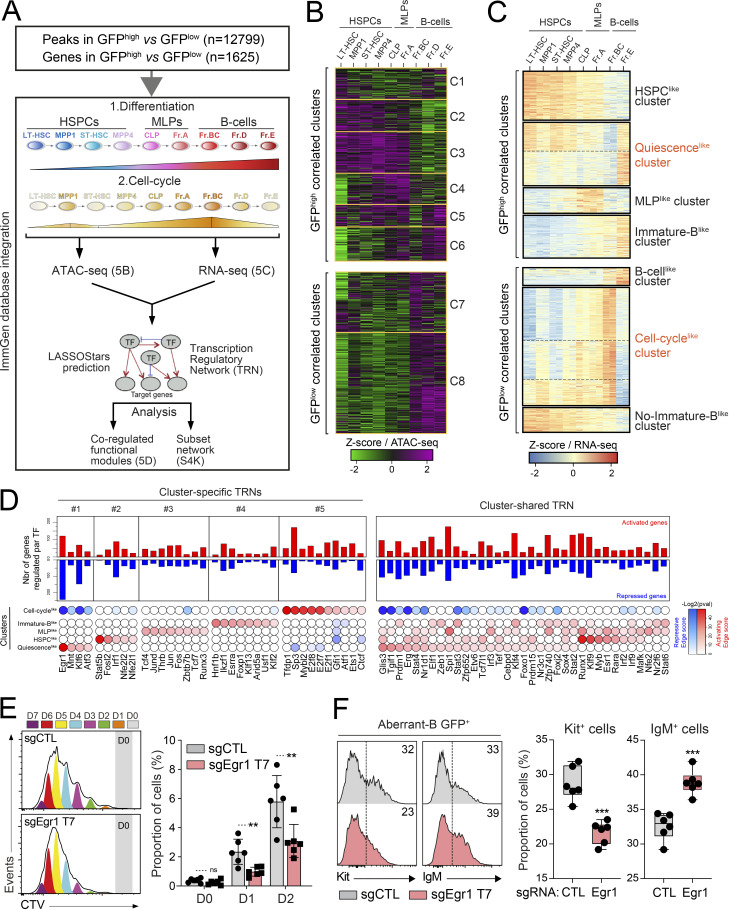
B cell acute lymphoblastic leukemia (B-ALL) is a multistep disease characterized by the hierarchical acquisition of genetic alterations. However, the question of how a primary oncogene reprograms stem cell-like properties in committed B cells and leads to a preneoplastic population remains unclear. Here, we used the PAX5::ELN oncogenic model to demonstrate a causal link between the differentiation blockade, the self-renewal, and the emergence of preleukemic stem cells (pre-LSCs). We show that PAX5::ELN disrupts the differentiation of preleukemic cells by enforcing the IL7r/JAK-STAT pathway. This disruption is associated with the induction of rare and quiescent pre-LSCs that sustain the leukemia-initiating activity, as assessed using the H2B-GFP model. Integration of transcriptomic and chromatin accessibility data reveals that those quiescent pre-LSCs lose B cell identity and reactivate an immature molecular program, reminiscent of human B-ALL chemo-resistant cells. Finally, our transcriptional regulatory network reveals the transcription factor EGR1 as a strong candidate to control quiescence/resistance of PAX5::ELN pre-LSCs as well as of blasts from human B-ALL.
© 2023 Fregona et al.
Conflict of interest statement
Disclosures: The authors declare no competing interests exist.
Figures










References
-
- Adelman, E.R., Huang H.T., Roisman A., Olsson A., Colaprico A., Qin T., Lindsley R.C., Bejar R., Salomonis N., Grimes H.L., and Figueroa M.E.. 2019. Aging human hematopoietic stem cells manifest profound epigenetic reprogramming of enhancers that may predispose to leukemia. Cancer Discov. 9:1080–1101. 10.1158/2159-8290.CD-18-1474 - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
