

Alveolar Differentiation Drives Resistance to KRAS Inhibition in Lung Adenocarcinoma
- PMID: 37931288
- PMCID: PMC10922405
- DOI: 10.1158/2159-8290.CD-23-0289
Alveolar Differentiation Drives Resistance to KRAS Inhibition in Lung Adenocarcinoma
Abstract
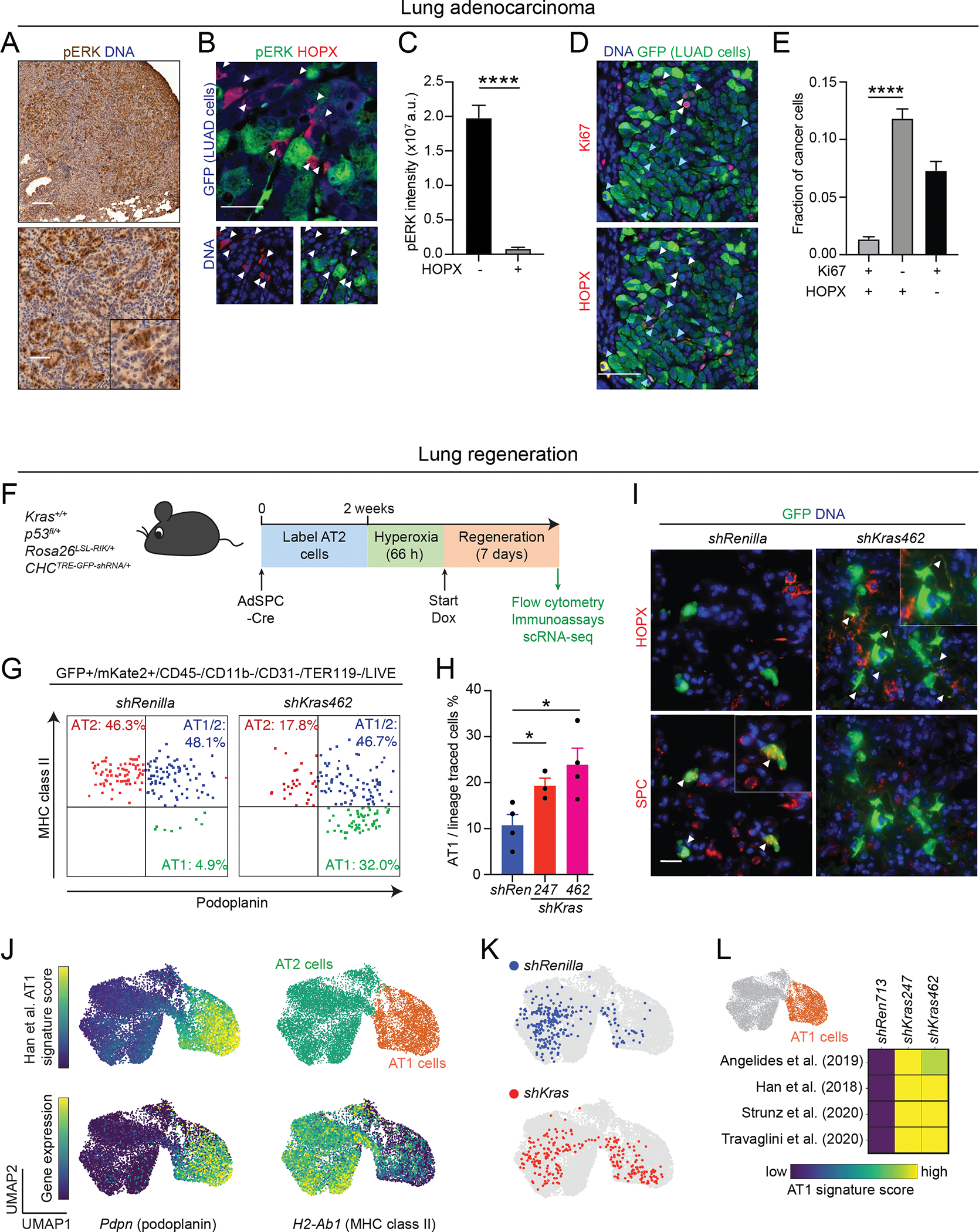
Lung adenocarcinoma (LUAD), commonly driven by KRAS mutations, is responsible for 7% of all cancer mortality. The first allele-specific KRAS inhibitors were recently approved in LUAD, but the clinical benefit is limited by intrinsic and acquired resistance. LUAD predominantly arises from alveolar type 2 (AT2) cells, which function as facultative alveolar stem cells by self-renewing and replacing alveolar type 1 (AT1) cells. Using genetically engineered mouse models, patient-derived xenografts, and patient samples, we found inhibition of KRAS promotes transition to a quiescent AT1-like cancer cell state in LUAD tumors. Similarly, suppressing Kras induced AT1 differentiation of wild-type AT2 cells upon lung injury. The AT1-like LUAD cells exhibited high growth and differentiation potential upon treatment cessation, whereas ablation of the AT1-like cells robustly improved treatment response to KRAS inhibitors. Our results uncover an unexpected role for KRAS in promoting intratumoral heterogeneity and suggest that targeting alveolar differentiation may augment KRAS-targeted therapies in LUAD.
Significance: Treatment resistance limits response to KRAS inhibitors in LUAD patients. We find LUAD residual disease following KRAS targeting is composed of AT1-like cancer cells with the capacity to reignite tumorigenesis. Targeting the AT1-like cells augments responses to KRAS inhibition, elucidating a therapeutic strategy to overcome resistance to KRAS-targeted therapy. This article is featured in Selected Articles from This Issue, p. 201.
©2023 American Association for Cancer Research.
Figures





Update of
-
Alveolar differentiation drives resistance to KRAS inhibition in lung adenocarcinoma.bioRxiv [Preprint]. 2023 Oct 1:2023.09.29.560194. doi: 10.1101/2023.09.29.560194. bioRxiv. 2023. Update in: Cancer Discov. 2024 Feb 8;14(2):308-325. doi: 10.1158/2159-8290.CD-23-0289. PMID: 37808711 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
