

Genetics and epidemiology of mutational barcode-defined clonal hematopoiesis
- PMID: 37932435
- PMCID: PMC10703693
- DOI: 10.1038/s41588-023-01555-z
Genetics and epidemiology of mutational barcode-defined clonal hematopoiesis
Abstract
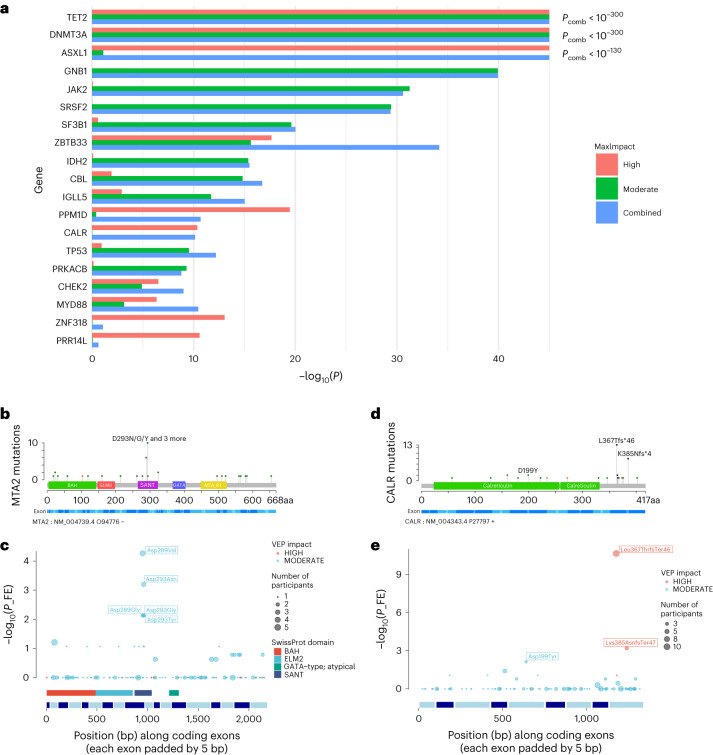
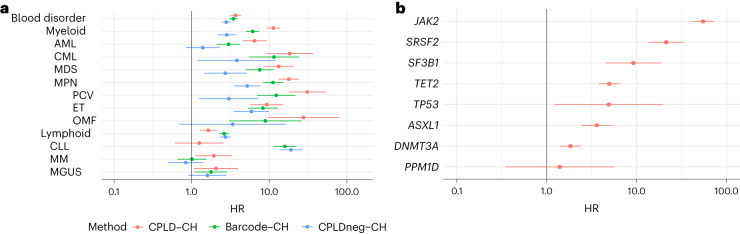
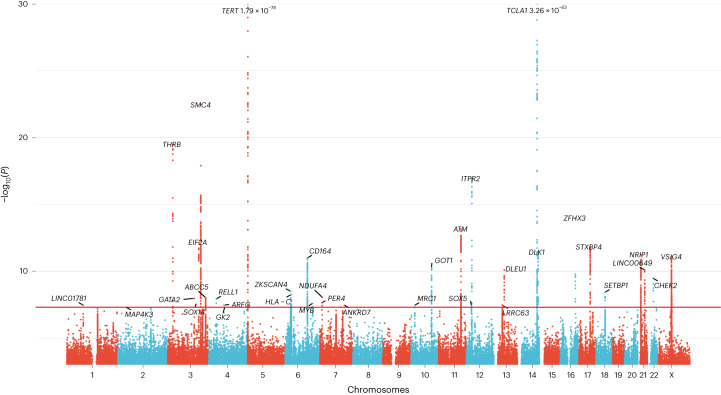
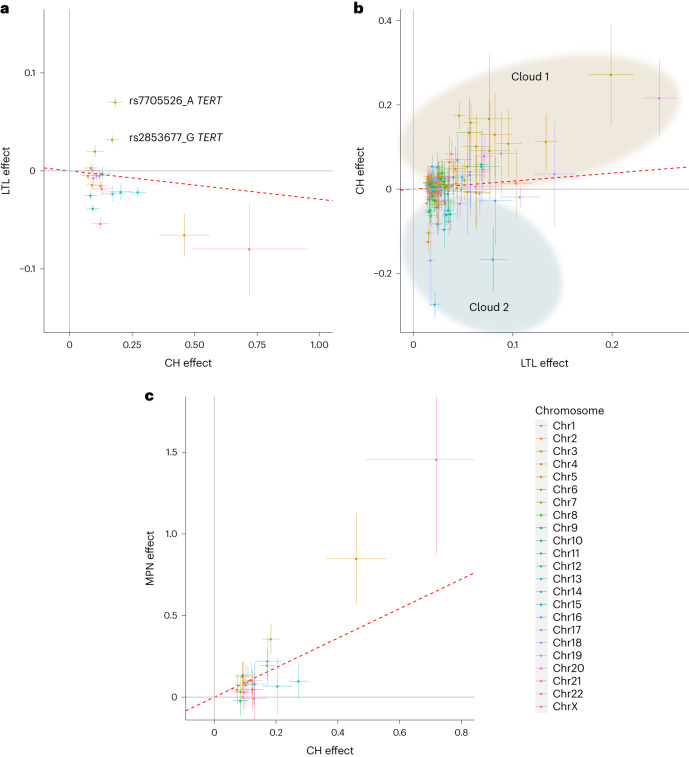
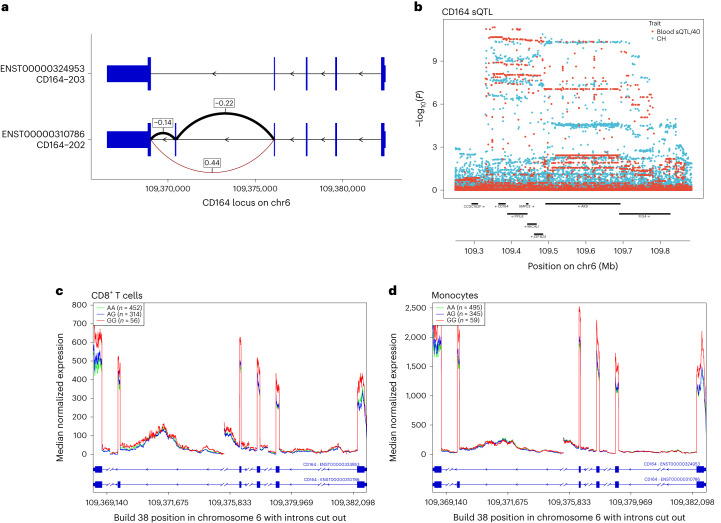
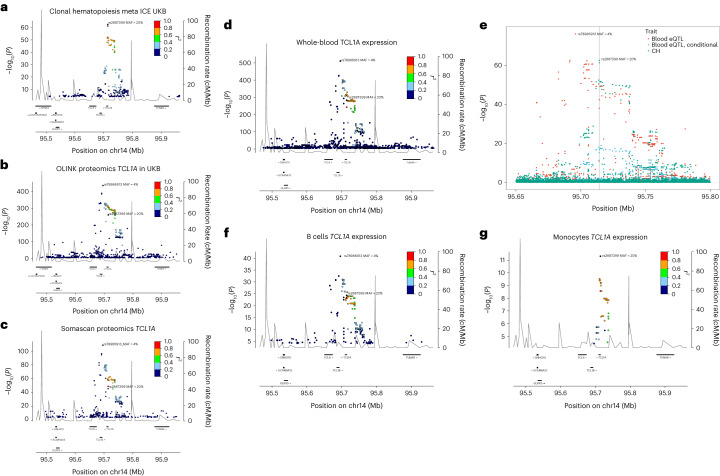
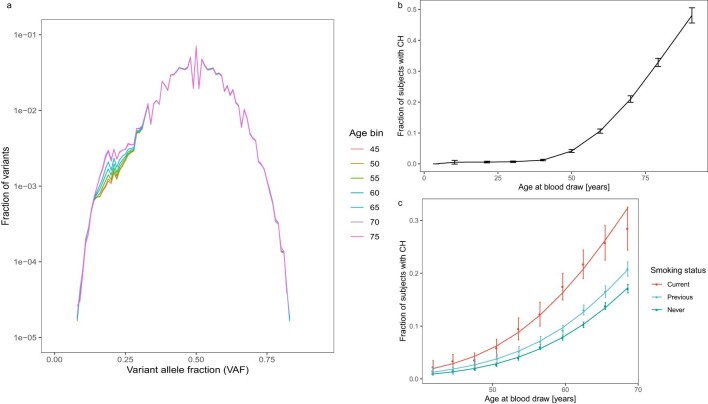
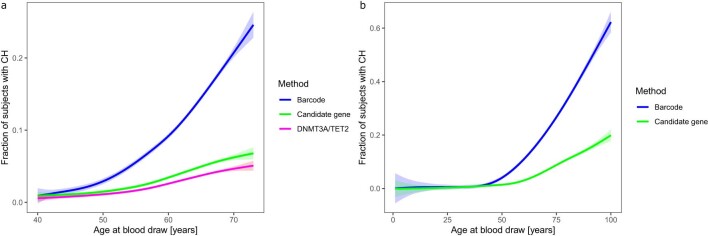
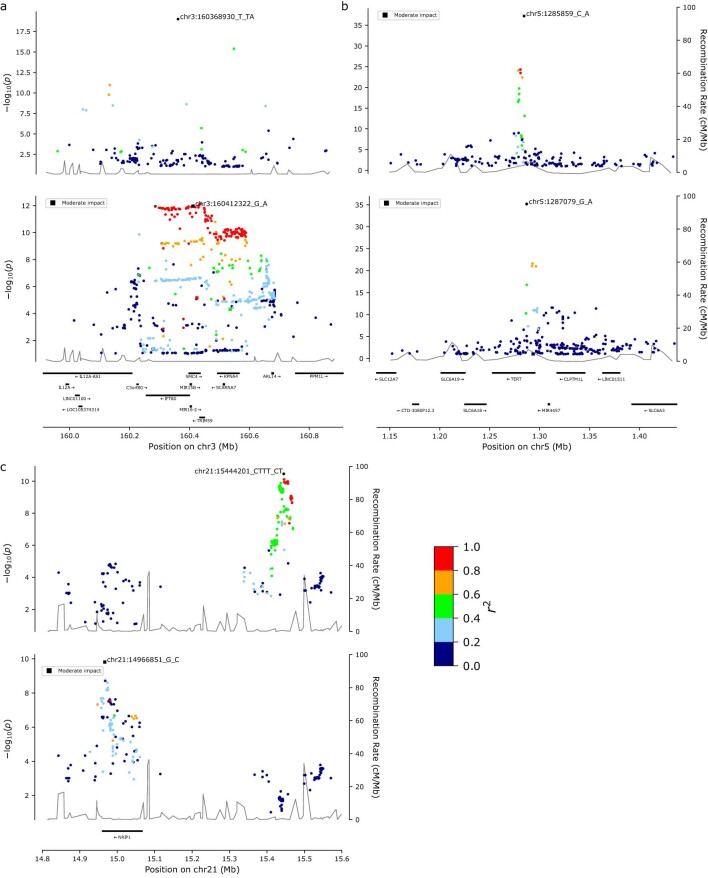
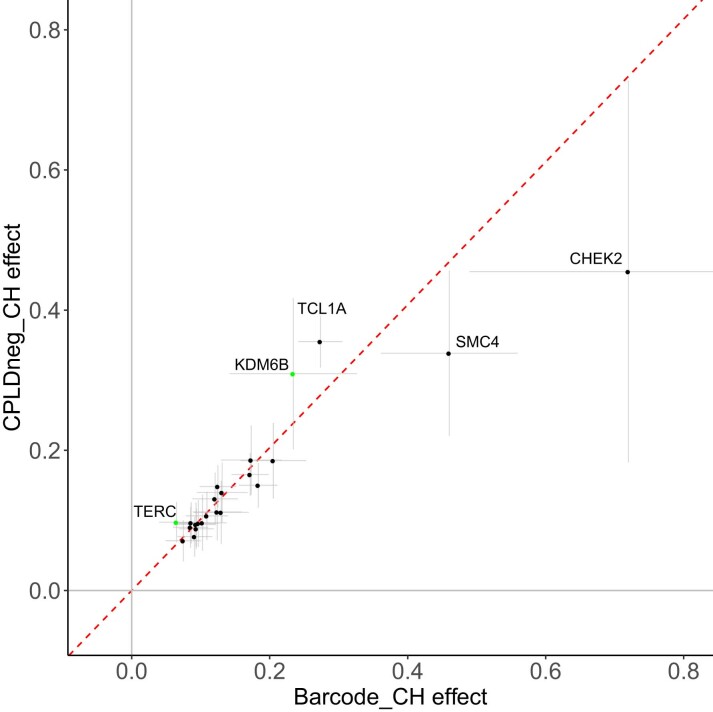
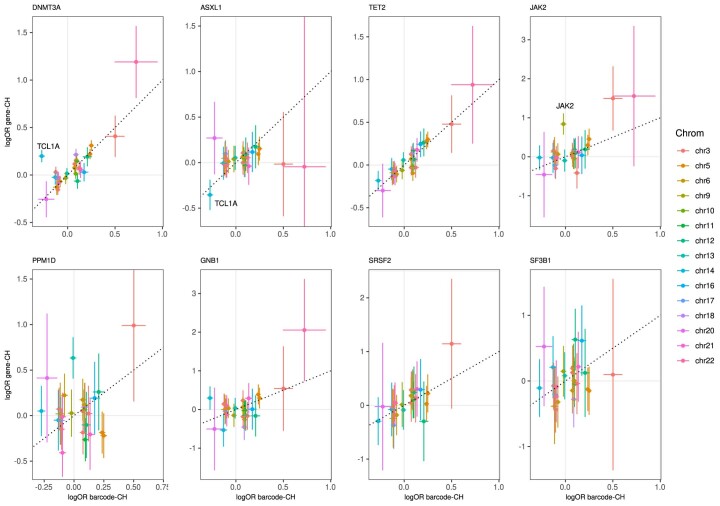
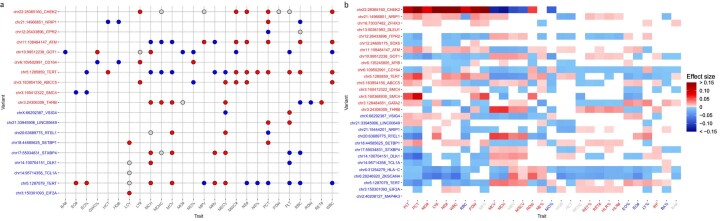
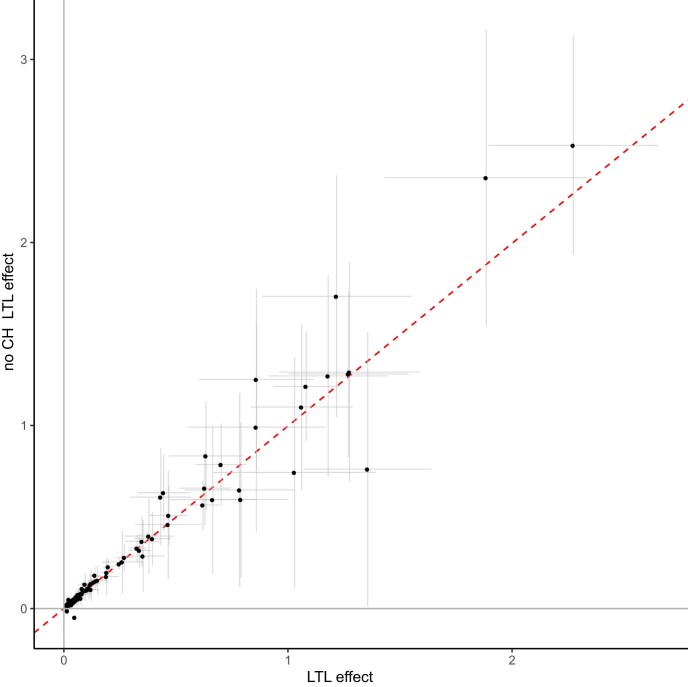
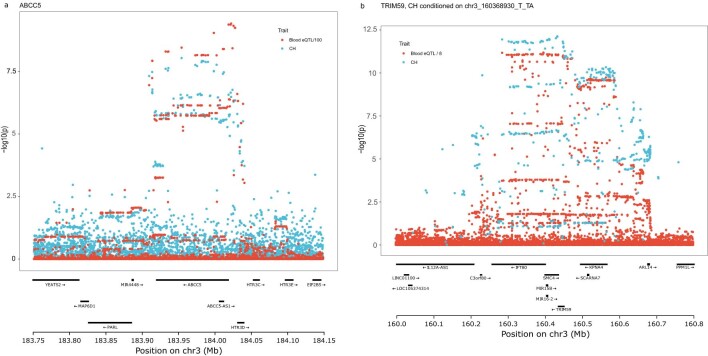
Clonal hematopoiesis (CH) arises when a substantial proportion of mature blood cells is derived from a single hematopoietic stem cell lineage. Using whole-genome sequencing of 45,510 Icelandic and 130,709 UK Biobank participants combined with a mutational barcode method, we identified 16,306 people with CH. Prevalence approaches 50% in elderly participants. Smoking demonstrates a dosage-dependent impact on risk of CH. CH associates with several smoking-related diseases. Contrary to published claims, we find no evidence that CH is associated with cardiovascular disease. We provide evidence that CH is driven by genes that are commonly mutated in myeloid neoplasia and implicate several new driver genes. The presence and nature of a driver mutation alters the risk profile for hematological disorders. Nevertheless, most CH cases have no known driver mutations. A CH genome-wide association study identified 25 loci, including 19 not implicated previously in CH. Splicing, protein and expression quantitative trait loci were identified for CD164 and TCL1A.
© 2023. The Author(s).
Conflict of interest statement
All deCODE authors are employees of the biotechnology company deCODE genetics/Amgen. The remaining authors declare no competing interests.
Figures
References
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
