

Targeting FLT3-TAZ signaling to suppress drug resistance in blast phase chronic myeloid leukemia
- PMID: 37932786
- PMCID: PMC10626670
- DOI: 10.1186/s12943-023-01837-4
Targeting FLT3-TAZ signaling to suppress drug resistance in blast phase chronic myeloid leukemia
Abstract
Background: Although the development of BCR::ABL1 tyrosine kinase inhibitors (TKIs) rendered chronic myeloid leukemia (CML) a manageable condition, acquisition of drug resistance during blast phase (BP) progression remains a critical challenge. Here, we reposition FLT3, one of the most frequently mutated drivers of acute myeloid leukemia (AML), as a prognostic marker and therapeutic target of BP-CML.
Methods: We generated FLT3 expressing BCR::ABL1 TKI-resistant CML cells and enrolled phase-specific CML patient cohort to obtain unpaired and paired serial specimens and verify the role of FLT3 signaling in BP-CML patients. We performed multi-omics approaches in animal and patient studies to demonstrate the clinical feasibility of FLT3 as a viable target of BP-CML by establishing the (1) molecular mechanisms of FLT3-driven drug resistance, (2) diagnostic methods of FLT3 protein expression and localization, (3) association between FLT3 signaling and CML prognosis, and (4) therapeutic strategies to tackle FLT3+ CML patients.
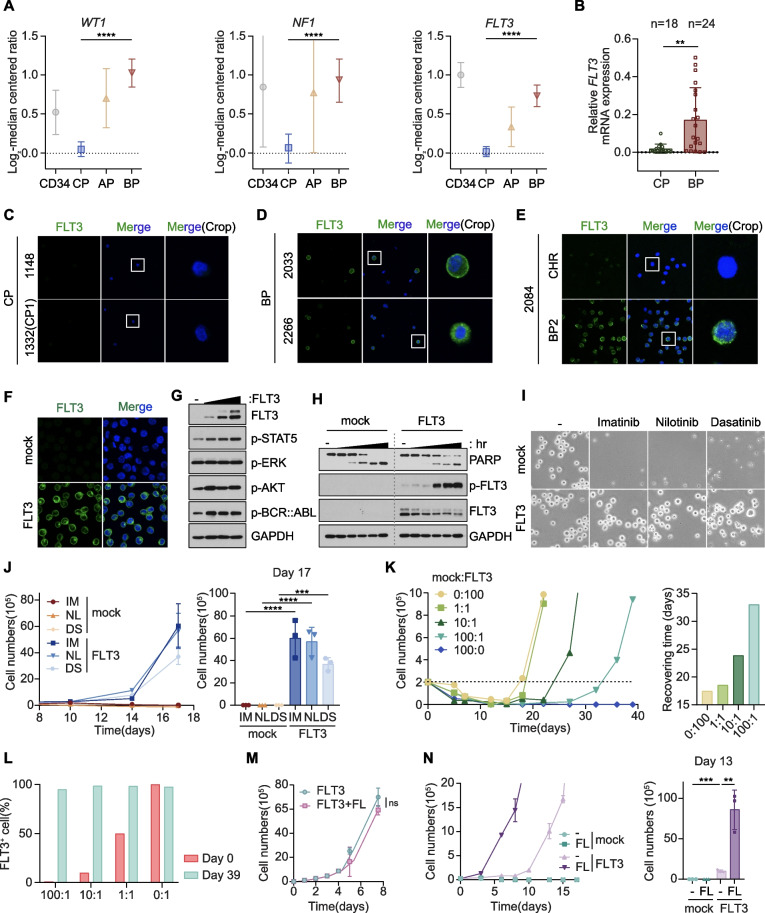
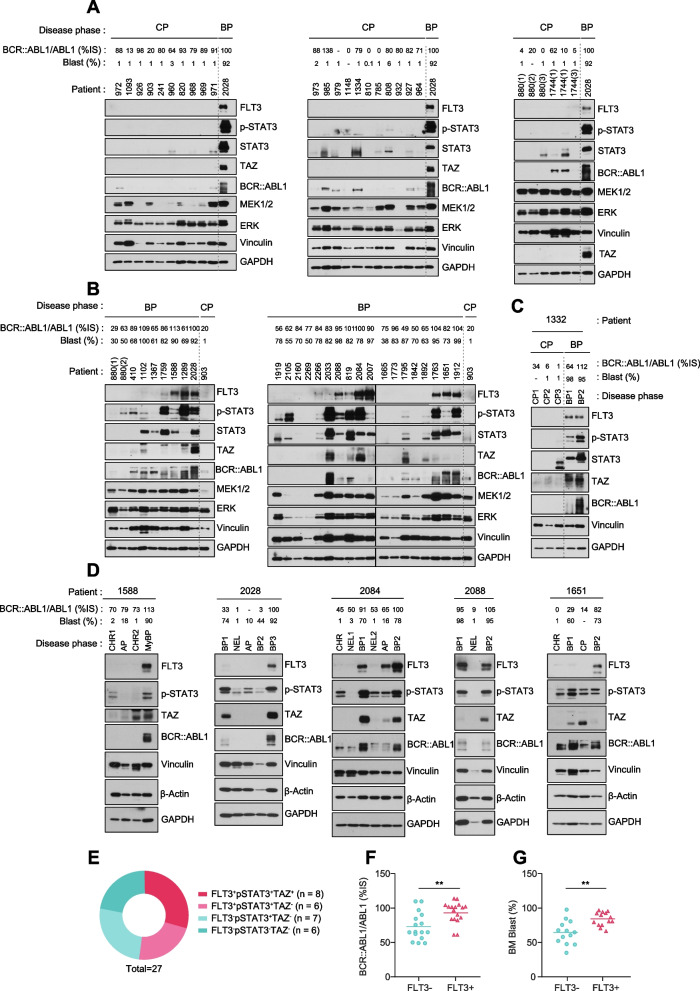
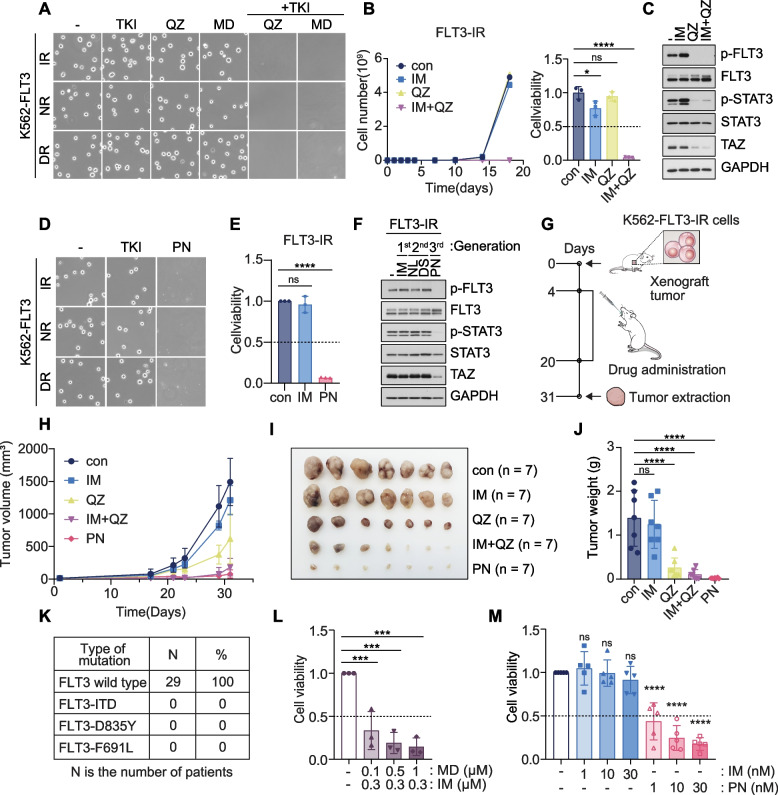
Results: We reposition the significance of FLT3 in the acquisition of drug resistance in BP-CML, thereby, newly classify a FLT3+ BP-CML subgroup. Mechanistically, FLT3 expression in CML cells activated the FLT3-JAK-STAT3-TAZ-TEAD-CD36 signaling pathway, which conferred resistance to a wide range of BCR::ABL1 TKIs that was independent of recurrent BCR::ABL1 mutations. Notably, FLT3+ BP-CML patients had significantly less favorable prognosis than FLT3- patients. Remarkably, we demonstrate that repurposing FLT3 inhibitors combined with BCR::ABL1 targeted therapies or the single treatment with ponatinib alone can overcome drug resistance and promote BP-CML cell death in patient-derived FLT3+ BCR::ABL1 cells and mouse xenograft models.
Conclusion: Here, we reposition FLT3 as a critical determinant of CML progression via FLT3-JAK-STAT3-TAZ-TEAD-CD36 signaling pathway that promotes TKI resistance and predicts worse prognosis in BP-CML patients. Our findings open novel therapeutic opportunities that exploit the undescribed link between distinct types of malignancies.
Keywords: AML; Blast phase; CD36; CML; Cancer; Drug resistance; FLT3; Hippo-YAP/TAZ pathway; Midostaurin; Ponatinib.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
