

TEAD Inhibition Overcomes YAP1/TAZ-Driven Primary and Acquired Resistance to KRASG12C Inhibitors
- PMID: 37934103
- PMCID: PMC10821578
- DOI: 10.1158/0008-5472.CAN-23-2994
TEAD Inhibition Overcomes YAP1/TAZ-Driven Primary and Acquired Resistance to KRASG12C Inhibitors
Abstract
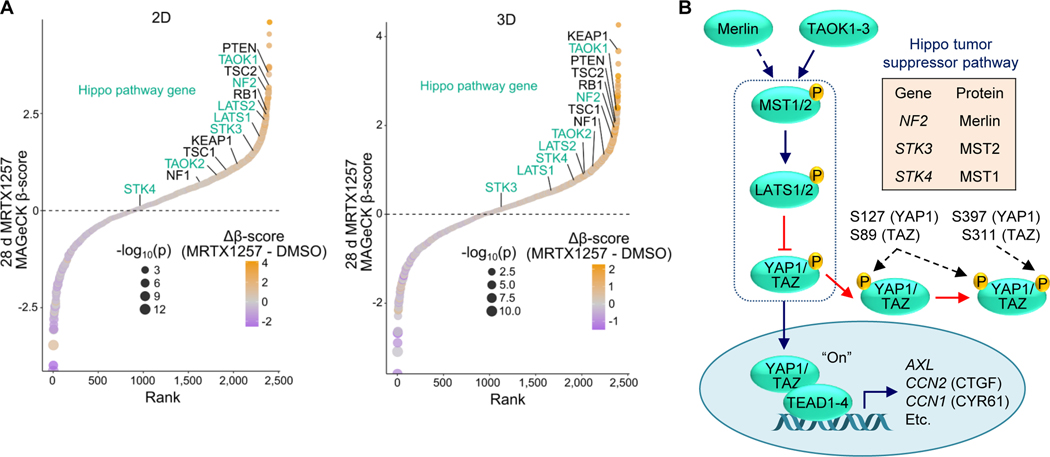
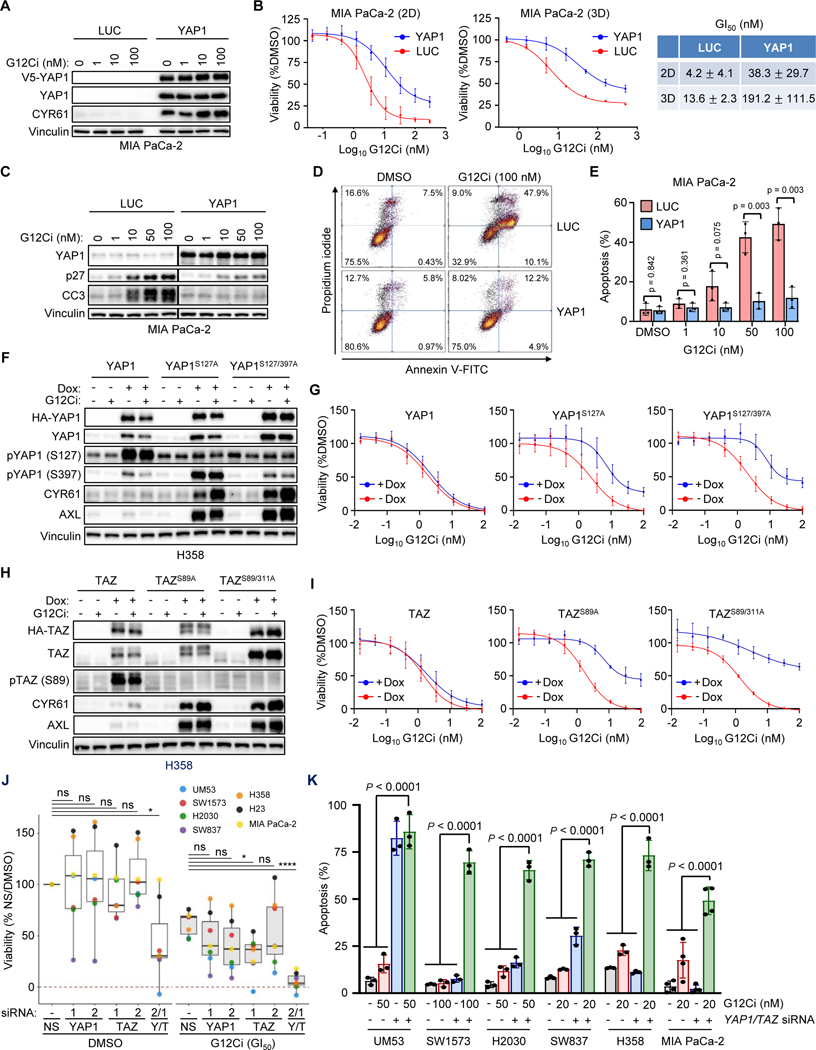
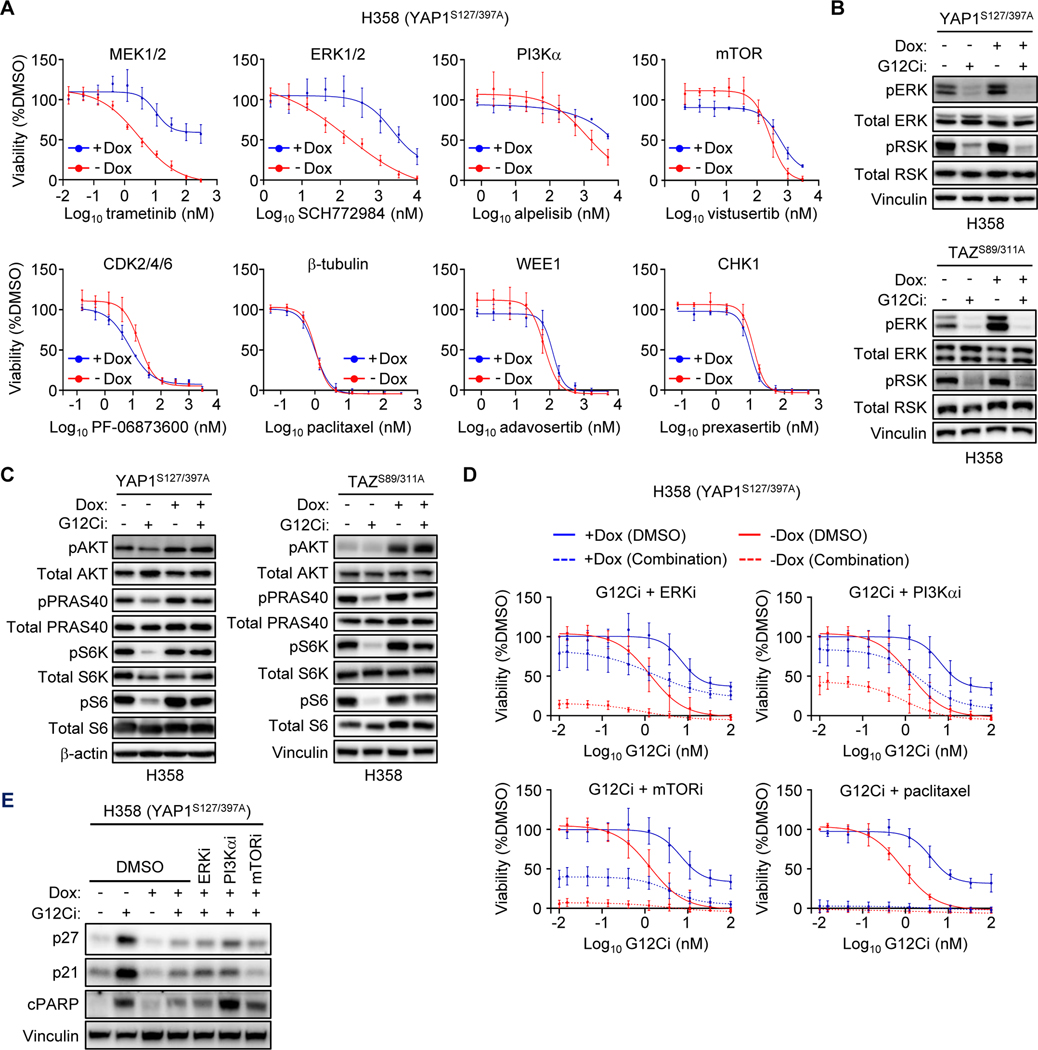
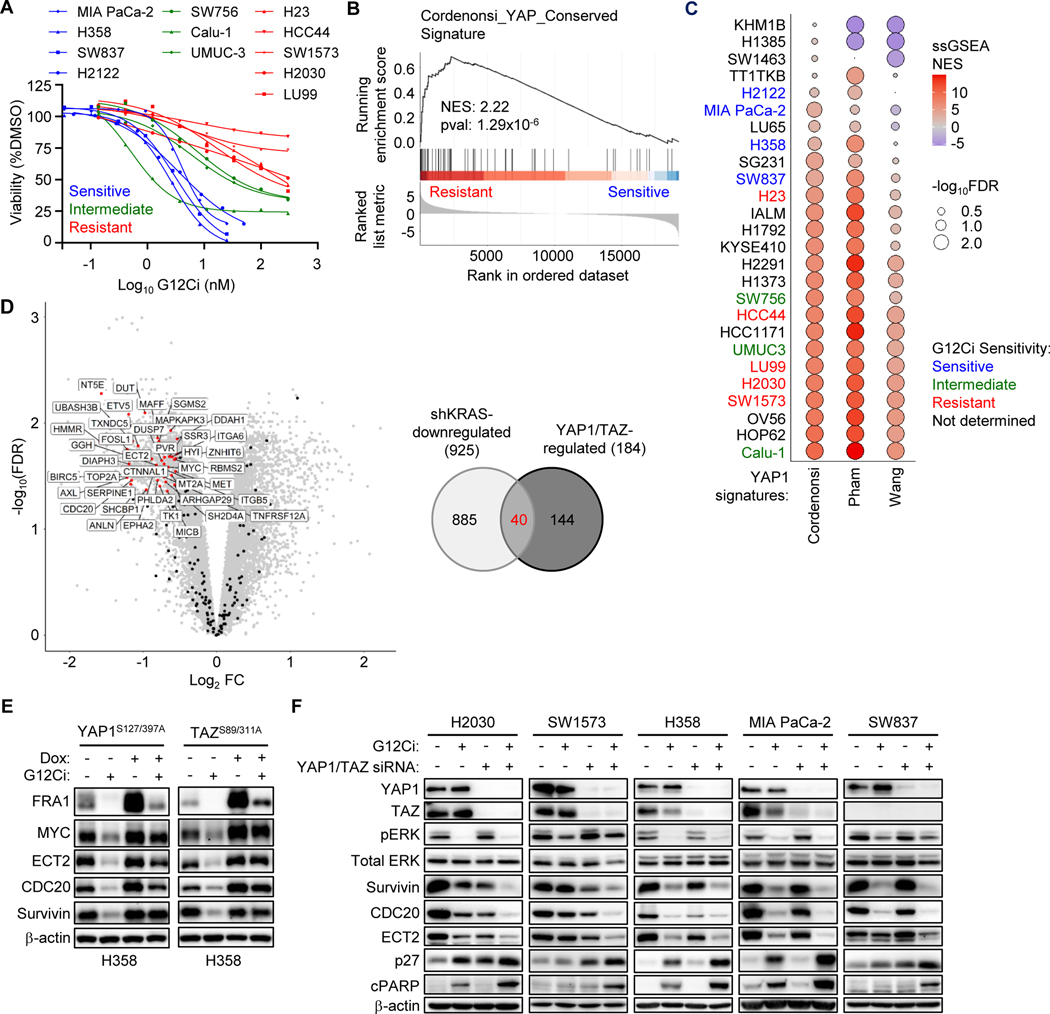
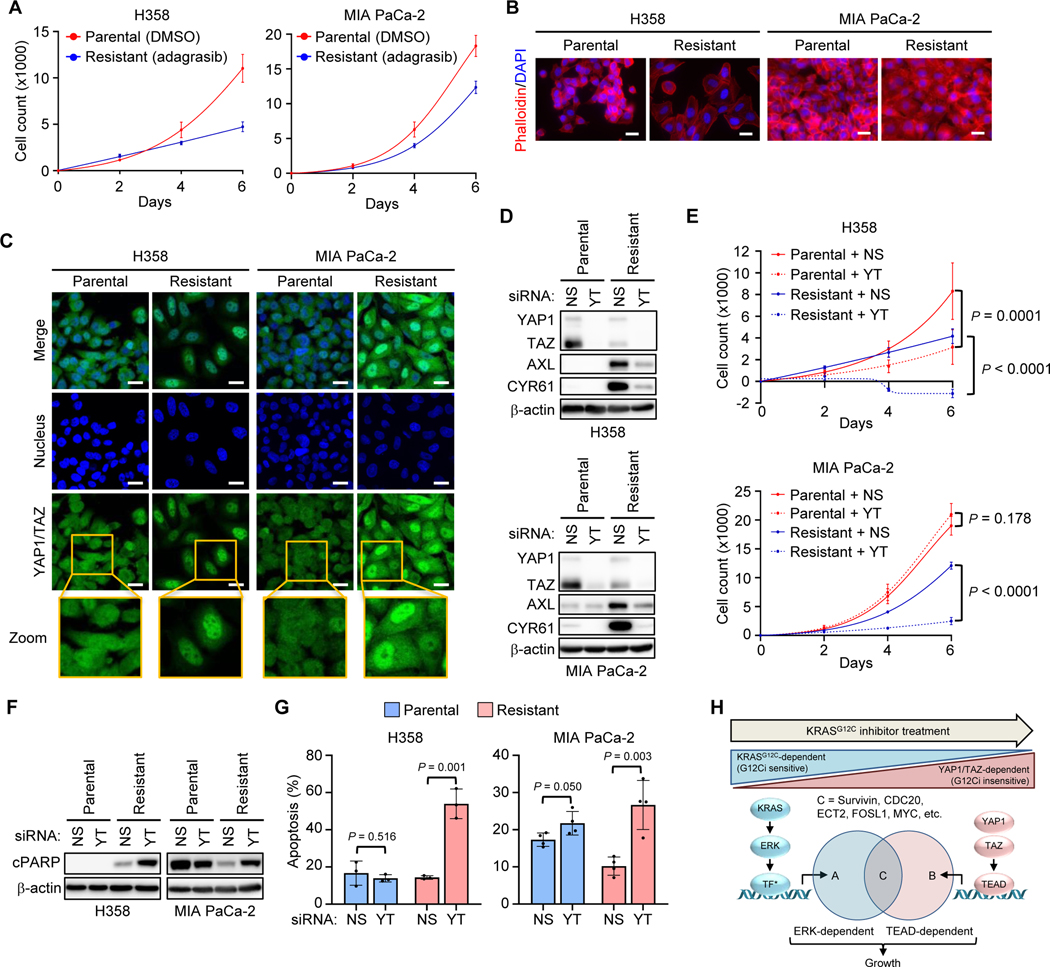
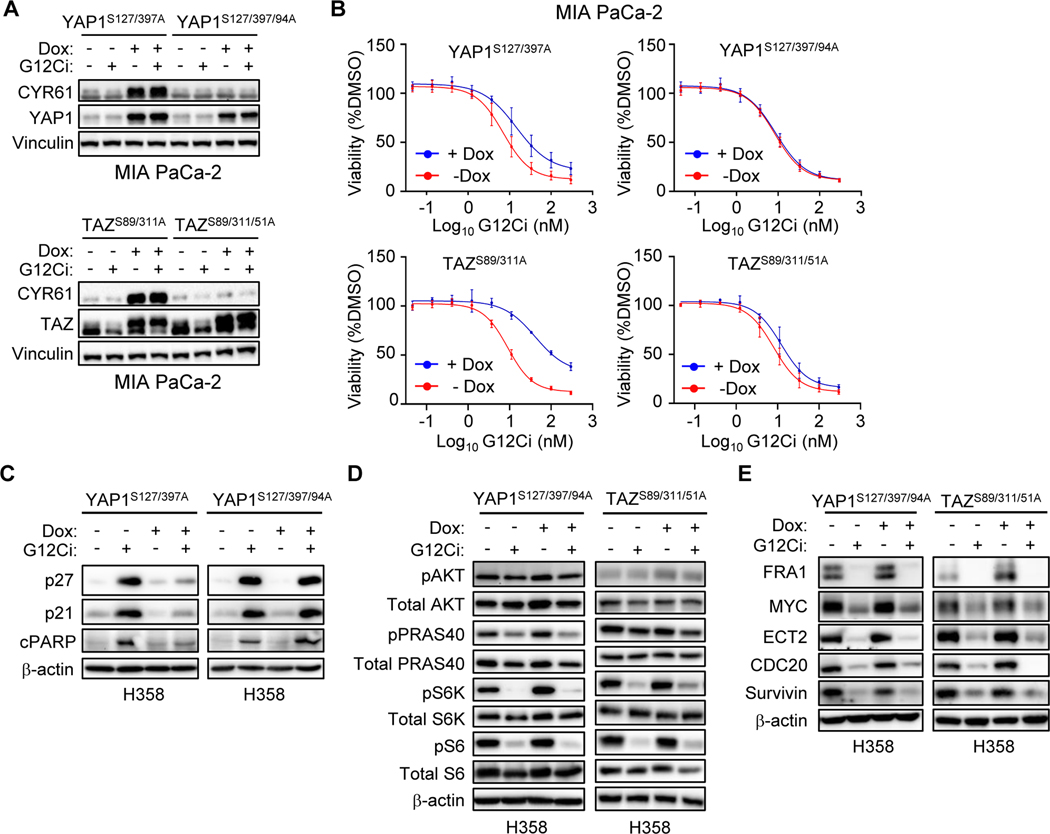
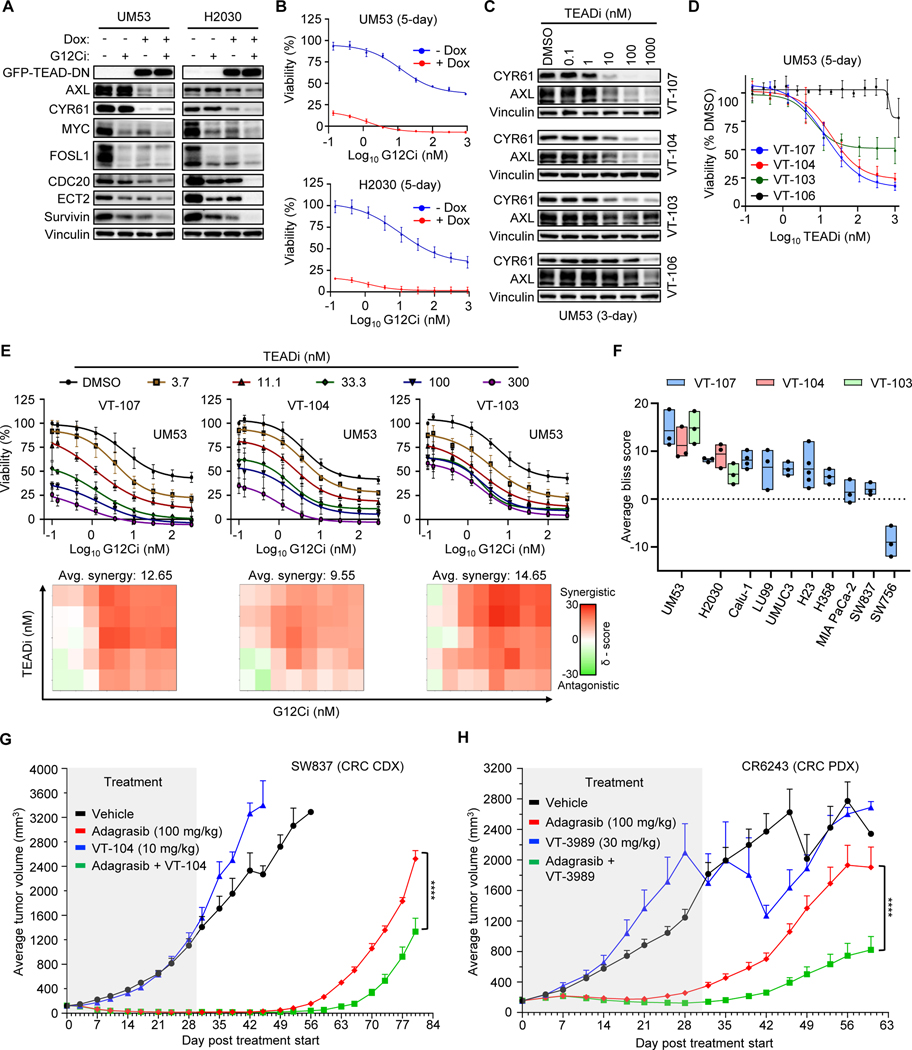
Primary/intrinsic and treatment-induced acquired resistance limit the initial response rate to and long-term efficacy of direct inhibitors of the KRASG12C mutant in cancer. To identify potential mechanisms of resistance, we applied a CRISPR/Cas9 loss-of-function screen and observed loss of multiple components of the Hippo tumor suppressor pathway, which acts to suppress YAP1/TAZ-regulated gene transcription. YAP1/TAZ activation impaired the antiproliferative and proapoptotic effects of KRASG12C inhibitor (G12Ci) treatment in KRASG12C-mutant cancer cell lines. Conversely, genetic suppression of YAP1/WWTR1 (TAZ) enhanced G12Ci sensitivity. YAP1/TAZ activity overcame KRAS dependency through two distinct TEAD transcription factor-dependent mechanisms, which phenocopy KRAS effector signaling. First, TEAD stimulated ERK-independent transcription of genes normally regulated by ERK (BIRC5, CDC20, ECT2, FOSL1, and MYC) to promote progression through the cell cycle. Second, TEAD caused activation of PI3K-AKT-mTOR signaling to overcome apoptosis. G12Ci treatment-induced acquired resistance was also caused by YAP1/TAZ-TEAD activation. Accordingly, concurrent treatment with pharmacologic inhibitors of TEAD synergistically enhanced KRASG12C inhibitor antitumor activity in vitro and prolonged tumor suppression in vivo. In summary, these observations reveal YAP1/TAZ-TEAD signaling as a crucial driver of primary and acquired resistance to KRAS inhibition and support the use of TEAD inhibitors to enhance the antitumor efficacy of KRAS-targeted therapies.
Significance: YAP1/TAZ-TEAD activation compensates for loss of KRAS effector signaling, establishing a mechanistic basis for concurrent inhibition of TEAD to enhance the efficacy of KRASG12C-selective inhibitor treatment of KRASG12C-mutant cancers. See related commentary by Johnson and Haigis, p. 4005.
©2023 American Association for Cancer Research.
Conflict of interest statement
Conflict of Interest
The other authors declare no competing interests.
Figures
Comment in
-
All Roads Lead to Rome: YAP/TAZ Activity Influences Efficacy of KRASG12C Inhibitors.Cancer Res. 2023 Dec 15;83(24):4005-4007. doi: 10.1158/0008-5472.CAN-23-3547. Cancer Res. 2023. PMID: 38098448
References
-
- Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin 2023;73:17–48. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
