

PGF2α signaling drives fibrotic remodeling and fibroblast population dynamics in mice
- PMID: 37934604
- PMCID: PMC10807712
- DOI: 10.1172/jci.insight.172977
PGF2α signaling drives fibrotic remodeling and fibroblast population dynamics in mice
Abstract
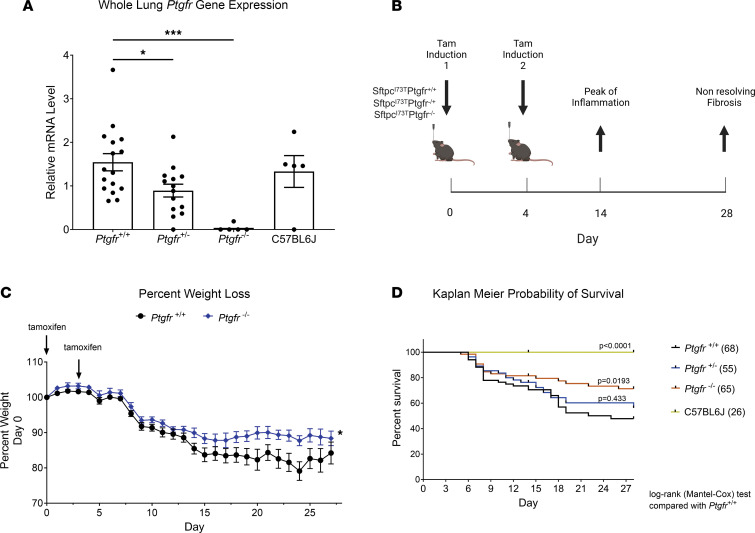
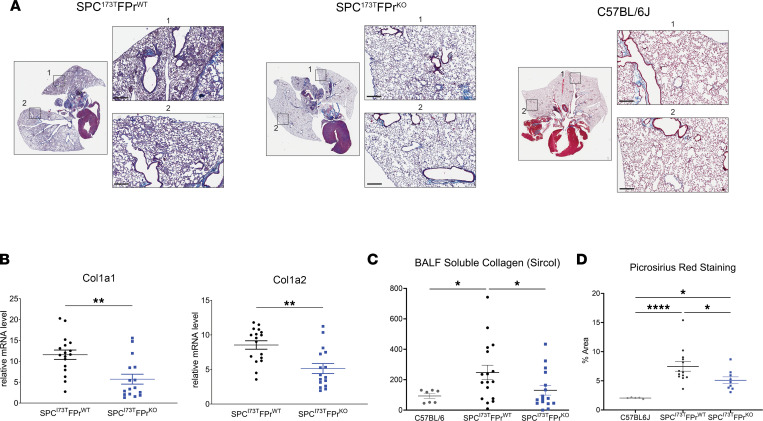
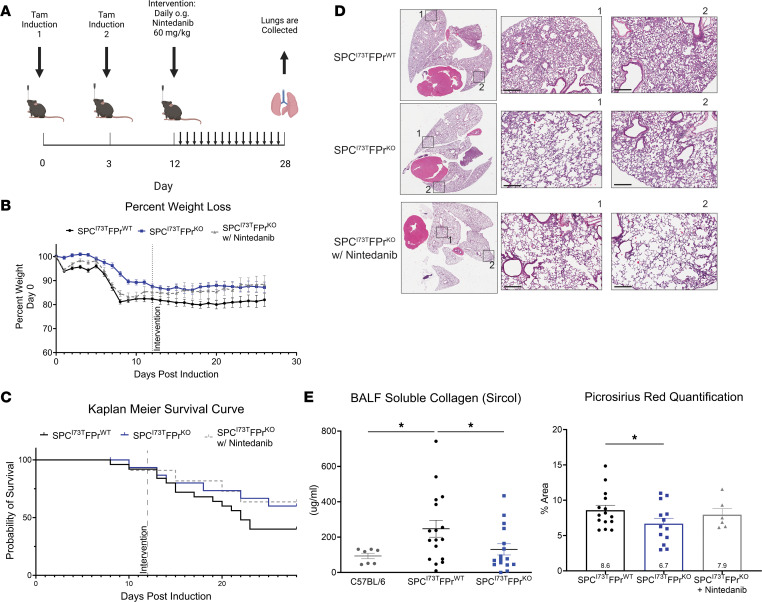
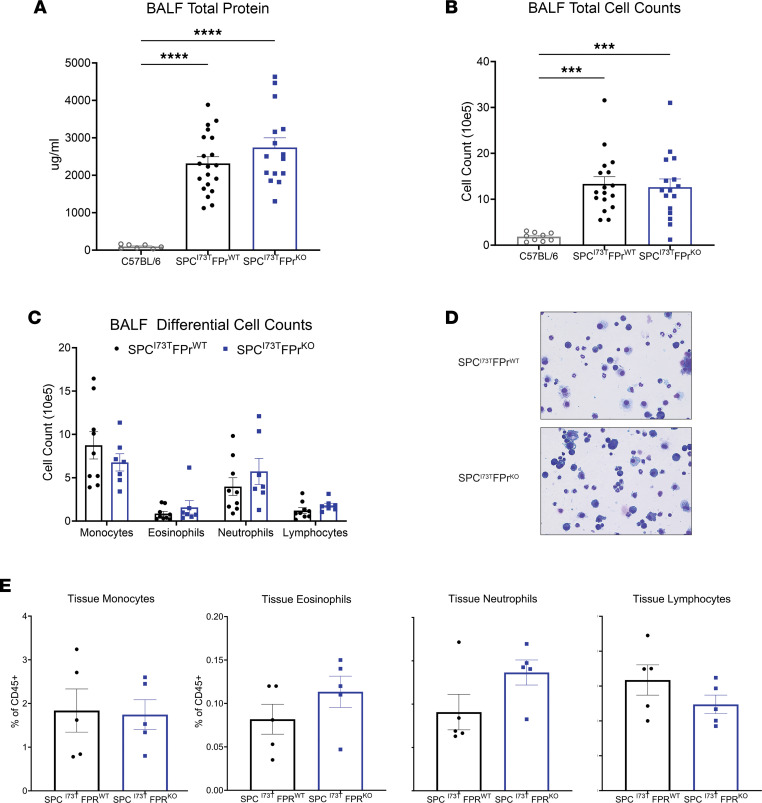
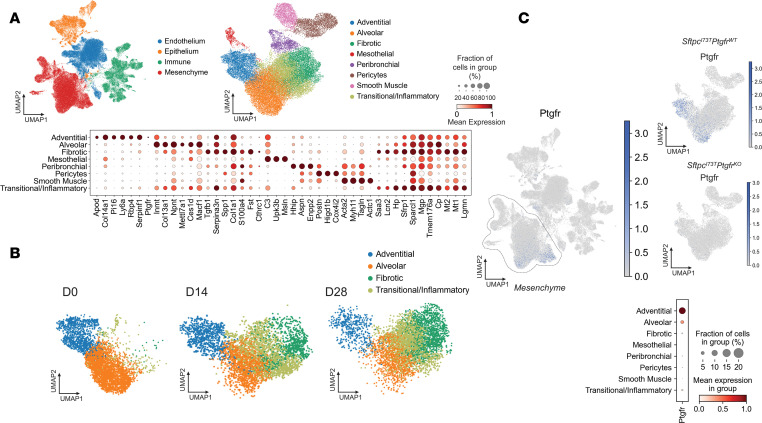
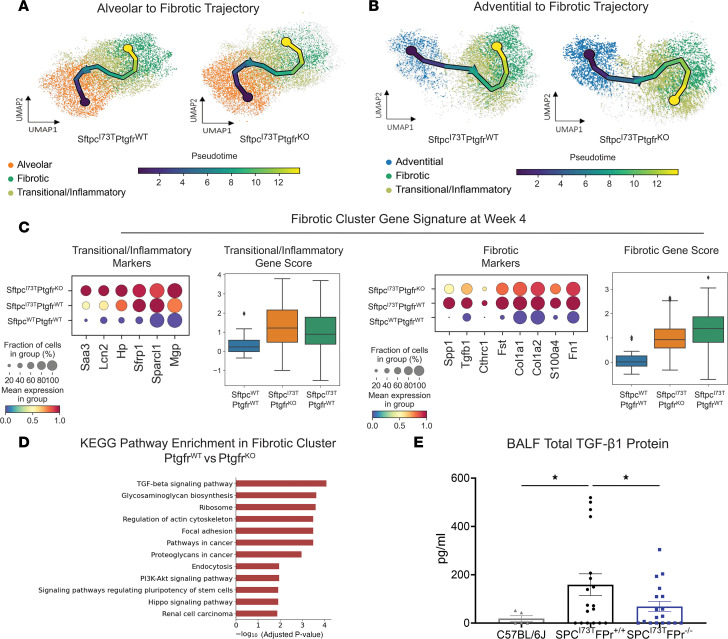
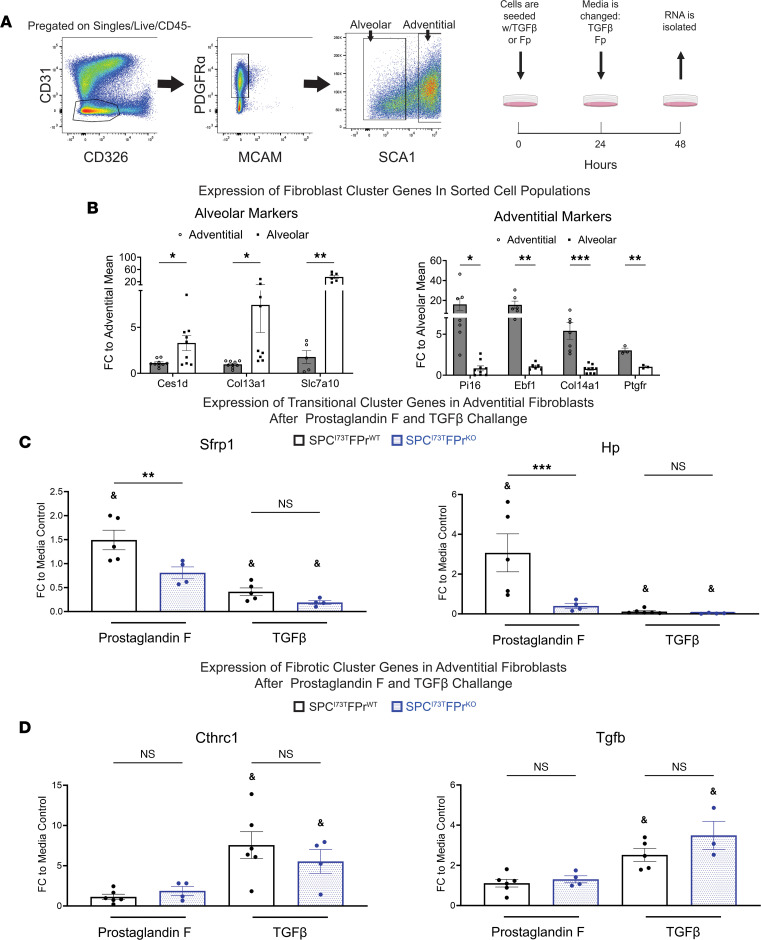
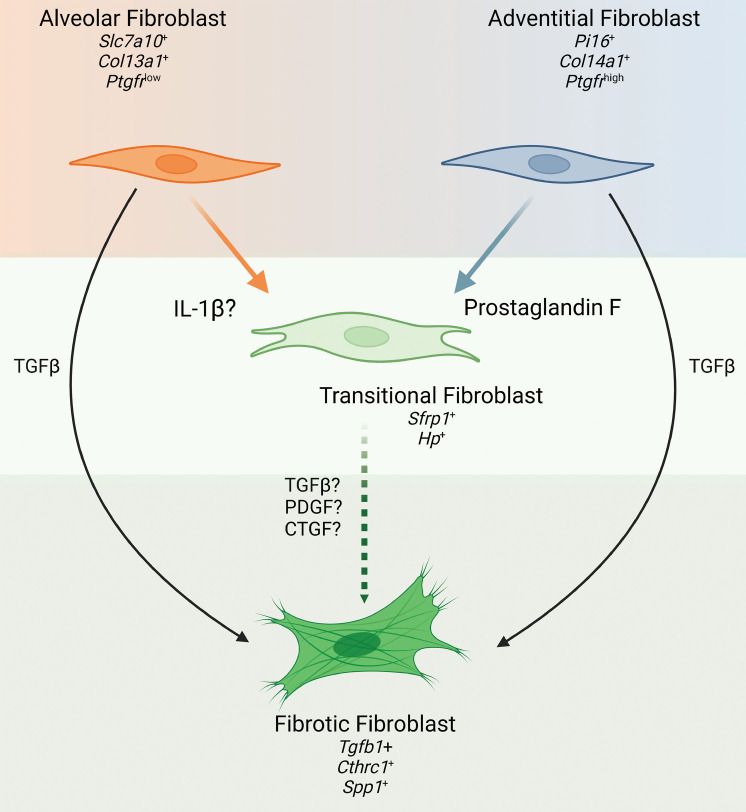
Idiopathic pulmonary fibrosis (IPF) is a chronic parenchymal lung disease characterized by repetitive alveolar cell injury, myofibroblast proliferation, and excessive extracellular matrix deposition for which unmet need persists for effective therapeutics. The bioactive eicosanoid, prostaglandin F2α, and its cognate receptor FPr (Ptgfr) are implicated as a TGF-β1-independent signaling hub for IPF. To assess this, we leveraged our published murine PF model (IER-SftpcI73T) expressing a disease-associated missense mutation in the surfactant protein C (Sftpc) gene. Tamoxifen-treated IER-SftpcI73T mice developed an early multiphasic alveolitis and transition to spontaneous fibrotic remodeling by 28 days. IER-SftpcI73T mice crossed to a Ptgfr-null (FPr-/-) line showed attenuated weight loss and gene dosage-dependent rescue of mortality compared with FPr+/+ cohorts. IER-SftpcI73T/FPr-/- mice also showed reductions in multiple fibrotic endpoints for which administration of nintedanib was not additive. Single-cell RNA-Seq, pseudotime analysis, and in vitro assays demonstrated Ptgfr expression predominantly within adventitial fibroblasts, which were reprogrammed to an "inflammatory/transitional" cell state in a PGF2α /FPr-dependent manner. Collectively, the findings provide evidence for a role for PGF2α signaling in IPF, mechanistically identify a susceptible fibroblast subpopulation, and establish a benchmark effect size for disruption of this pathway in mitigating fibrotic lung remodeling.
Keywords: Eicosanoids; Fibrosis; Molecular biology; Pulmonology.
Figures
Update of
-
Disruption of Prostaglandin F2α Receptor Signaling Attenuates Fibrotic Remodeling and Alters Fibroblast Population Dynamics in A Preclinical Murine Model of Idiopathic Pulmonary Fibrosis.bioRxiv [Preprint]. 2023 Jun 7:2023.06.07.543956. doi: 10.1101/2023.06.07.543956. bioRxiv. 2023. Update in: JCI Insight. 2023 Dec 22;8(24):e172977. doi: 10.1172/jci.insight.172977. PMID: 37333249 Free PMC article. Updated. Preprint.
References
-
- Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med. 2018;379(8):797–798. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
