

Spatiotemporal roles of AMPK in PARP-1- and autophagy-dependent retinal pigment epithelial cell death caused by UVA
- PMID: 37936170
- PMCID: PMC10629085
- DOI: 10.1186/s12929-023-00978-4
Spatiotemporal roles of AMPK in PARP-1- and autophagy-dependent retinal pigment epithelial cell death caused by UVA
Abstract
Background: Although stimulating autophagy caused by UV has been widely demonstrated in skin cells to exert cell protection, it remains unknown the cellular events in UVA-treated retinal pigment epithelial (RPE) cells.
Methods: Human ARPE-19 cells were used to measure cell viability, mitochondrial reactive oxygen species (ROS), mitochondrial membrane potential (MMP), mitochondrial mass and lysosomal mass by flow cytometry. Mitochondrial oxygen consumption rate (OCR) was recorded using Seahorse XF flux analyzer. Confocal microscopic images were performed to indicate the mitochondrial dynamics, LC3 level, and AMPK translocation after UVA irradiation.
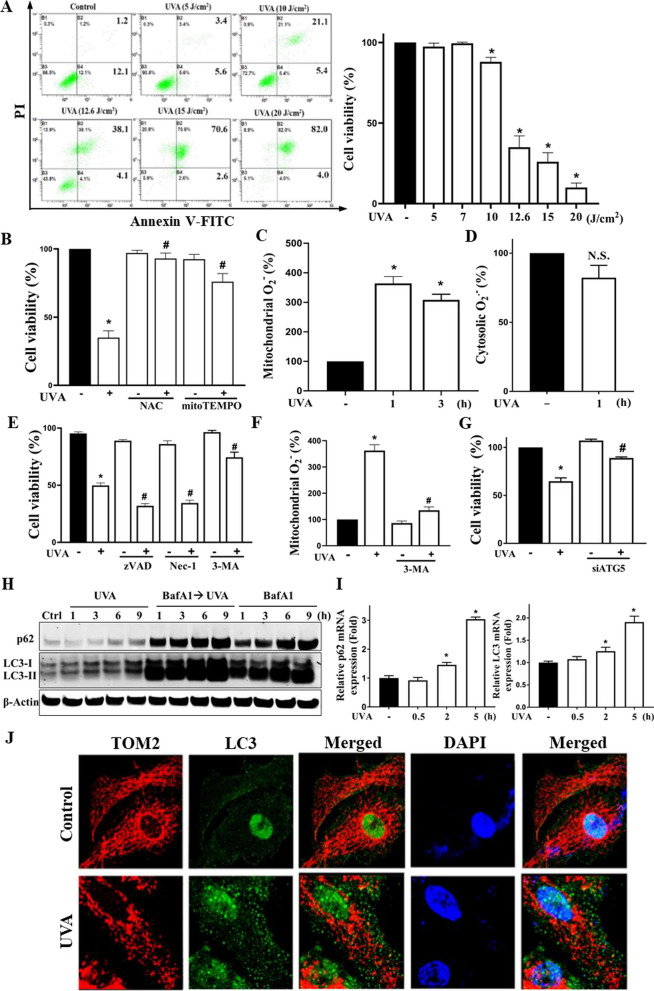
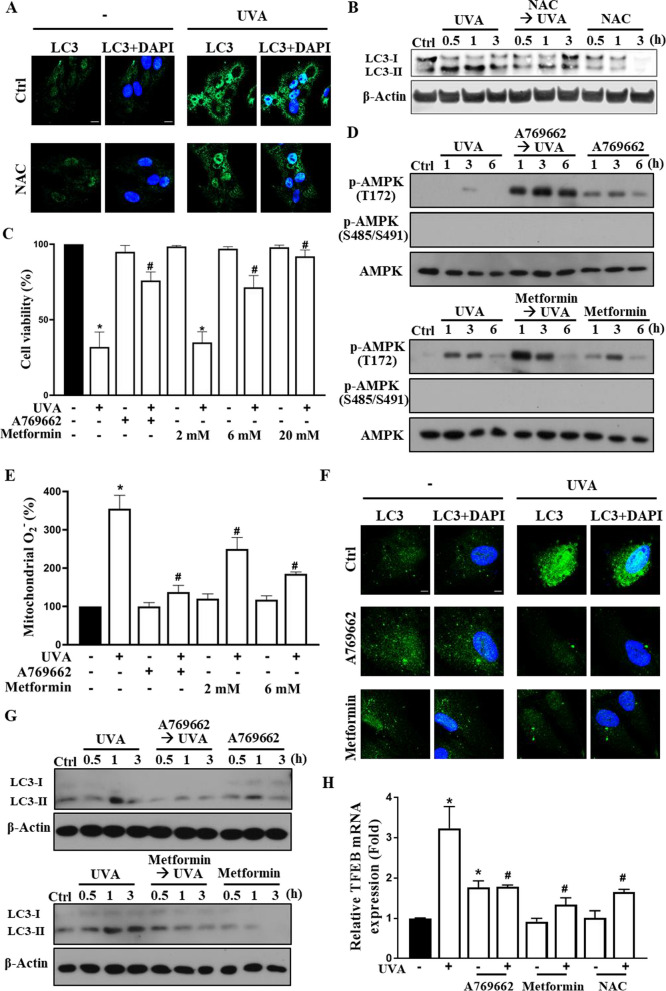
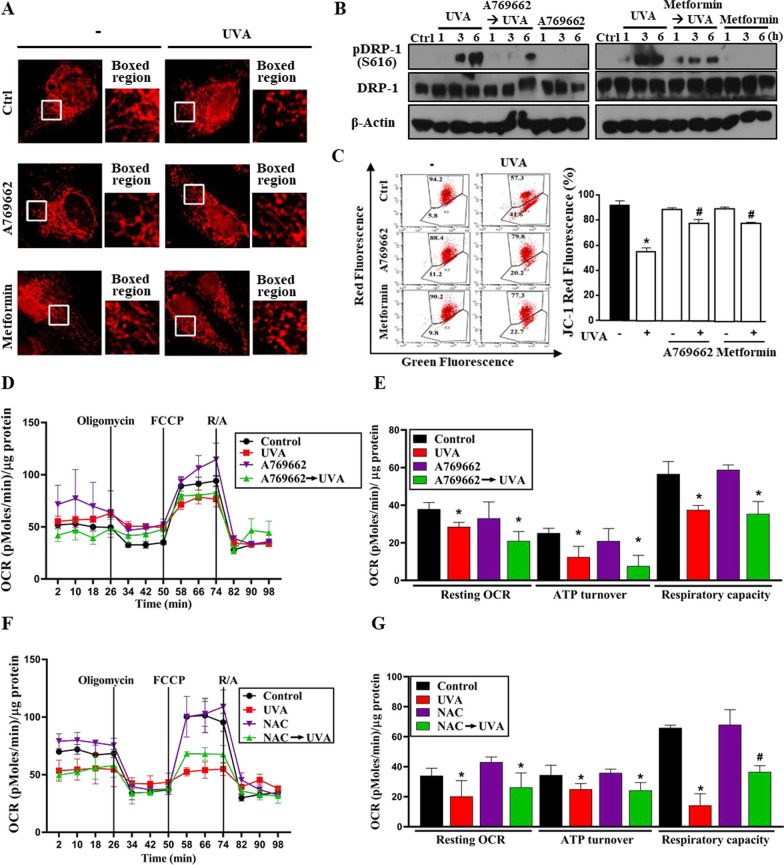
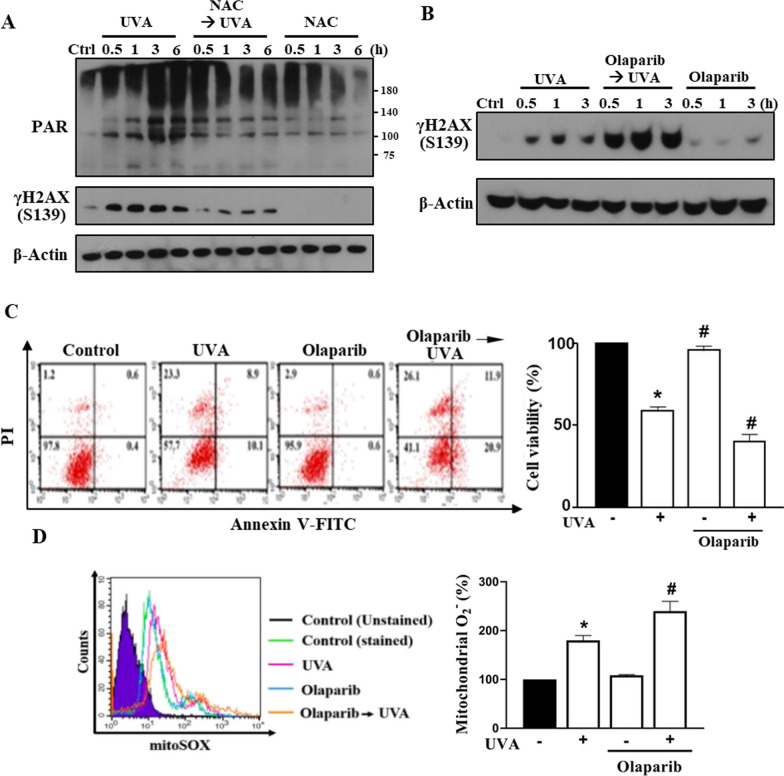
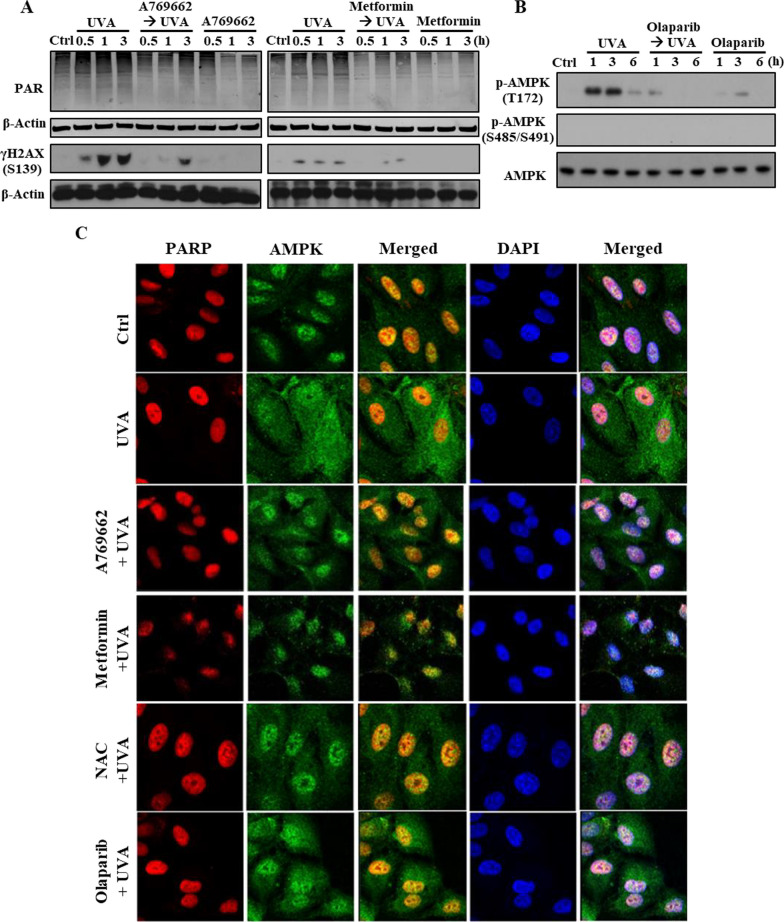
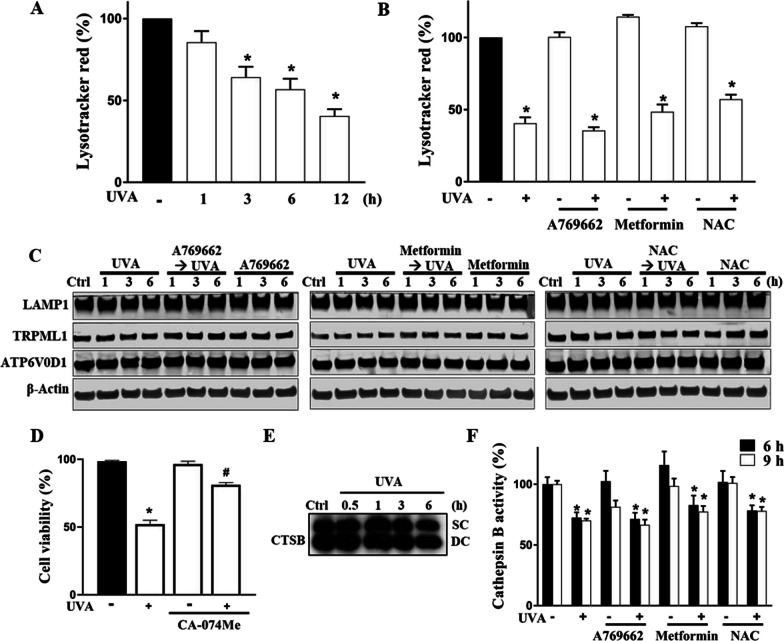
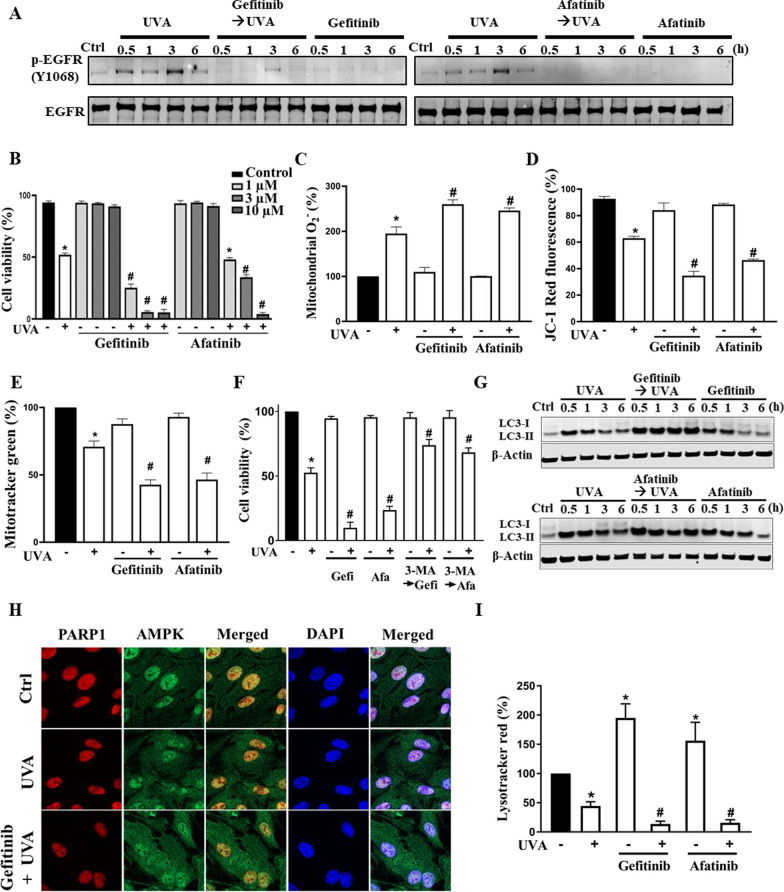
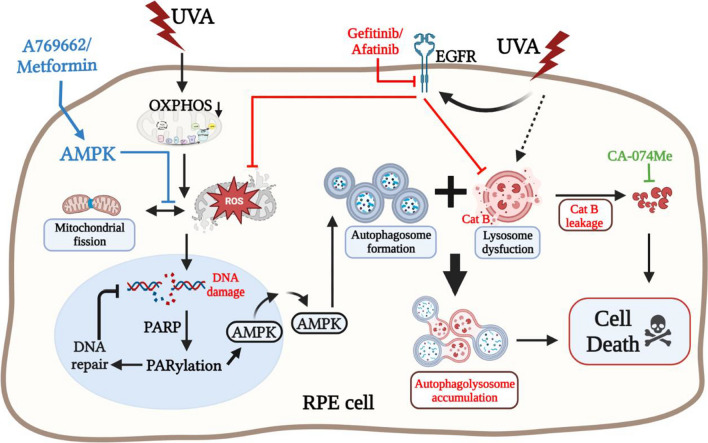
Results: We confirmed mitochondrial ROS production and DNA damage are two major features caused by UVA. We found the cell death is prevented by autophagy inhibitor 3-methyladenine and gene silencing of ATG5, and UVA induces ROS-dependent LC3II expression, LC3 punctate and TFEB expression, suggesting the autophagic death in the UVA-stressed RPE cells. Although PARP-1 inhibitor olaparib increases DNA damage, ROS production, and cell death, it also blocks AMPK activation caused by UVA. Interestingly we found a dramatic nuclear export of AMPK upon UVA irradiation which is blocked by N-acetylcysteine and olaparib. In addition, UVA exposure gradually decreases lysosomal mass and inhibits cathepsin B activity at late phase due to lysosomal dysfunction. Nevertheless, cathepsin B inhibitor, CA-074Me, reverses the death extent, suggesting the contribution of cathepsin B in the death pathway. When examining the role of EGFR in cellular events caused by UVA, we found that UVA can rapidly transactivate EGFR, and treatment with EGFR TKIs (gefitinib and afatinib) enhances the cell death accompanied by the increased LC3II formation, ROS production, loss of MMP and mass of mitochondria and lysosomes. Although AMPK activation by ROS-PARP-1 mediates autophagic cell death, we surprisingly found that pretreatment of cells with AMPK activators (A769662 and metformin) reverses cell death. Concomitantly, both agents block UVA-induced mitochondrial ROS production, autophagic flux, and mitochondrial fission without changing the inhibition of cathepsin B.
Conclusion: UVA exposure rapidly induces ROS-PARP-1-AMPK-autophagic flux and late lysosomal dysfunction. Pre-inducing AMPK activation can prevent cellular events caused by UVA and provide a new protective strategy in photo-oxidative stress and photo-retinopathy.
Keywords: AMPK; Autophagic cell death; EGFR; Lysosome dysfunction; PARP; ROS; RPE; UVA.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
