

Targeting of intracellular oncoproteins with peptide-centric CARs
- PMID: 37938771
- PMCID: PMC10665195
- DOI: 10.1038/s41586-023-06706-0
Targeting of intracellular oncoproteins with peptide-centric CARs
Abstract
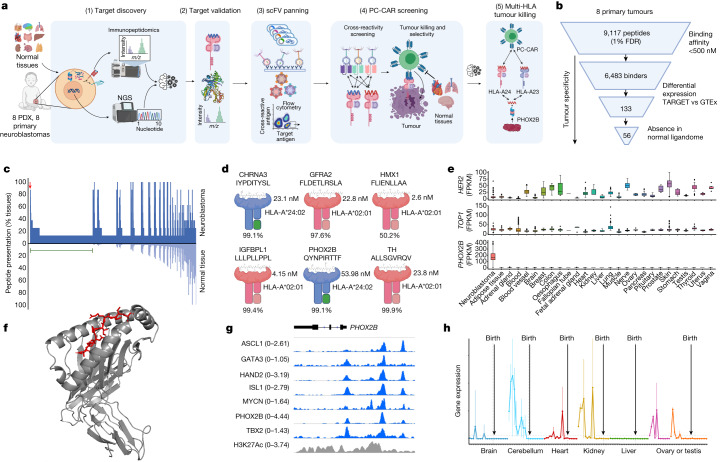
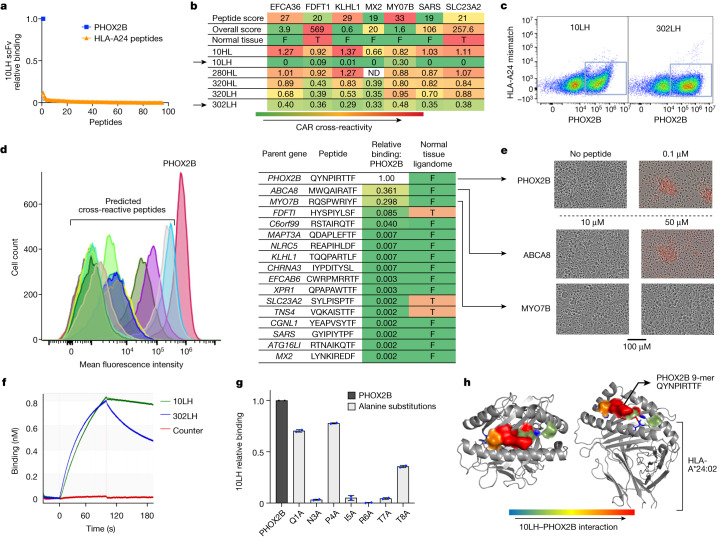
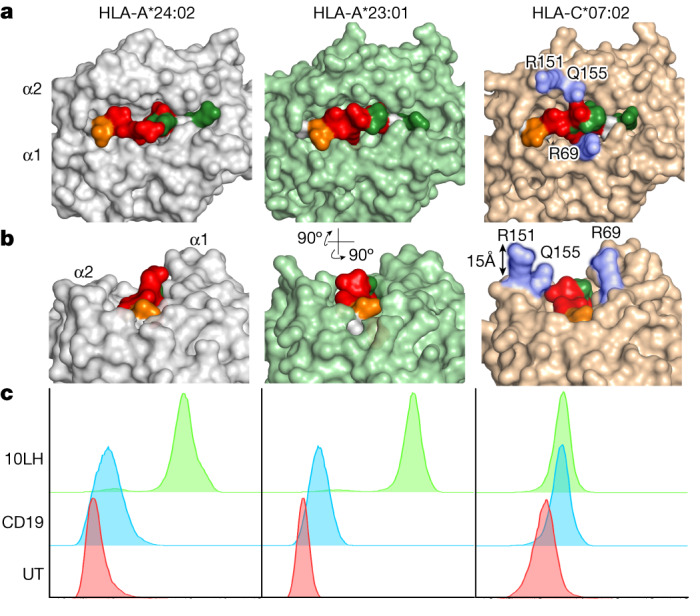
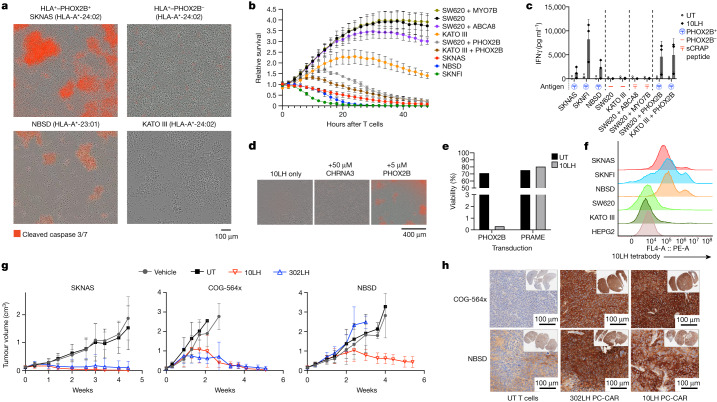
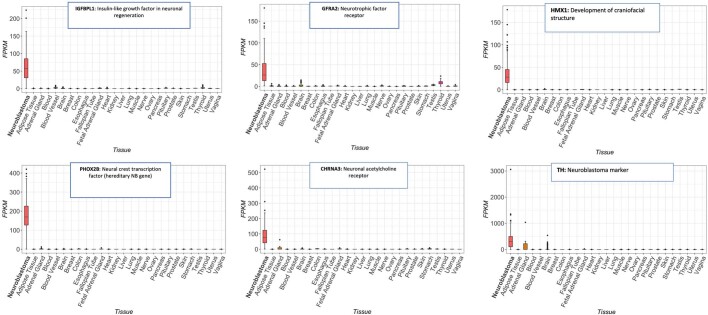
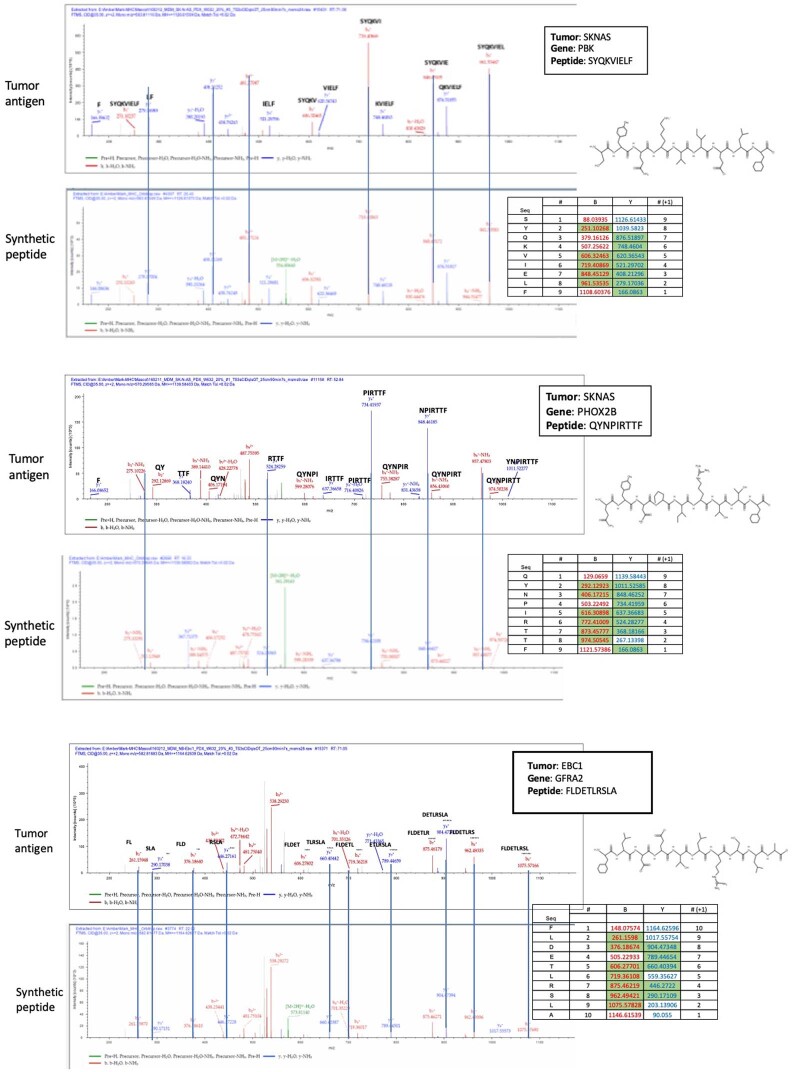
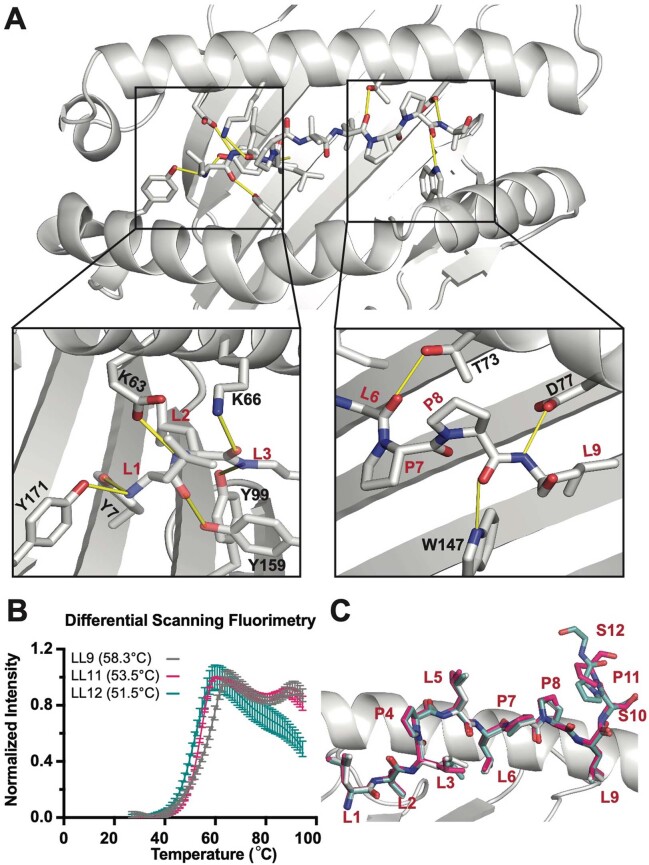
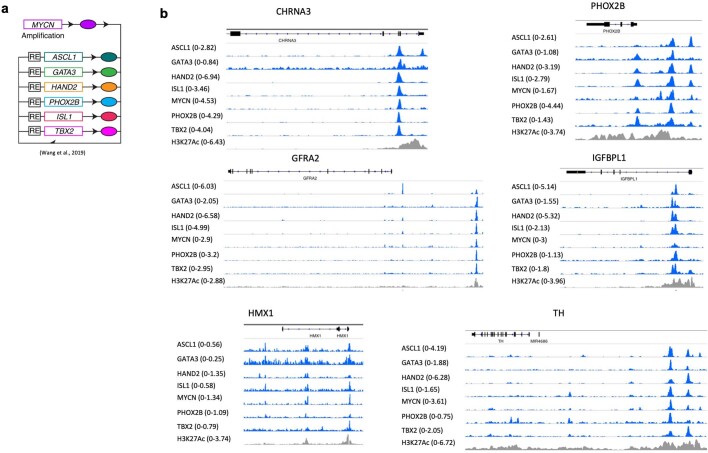
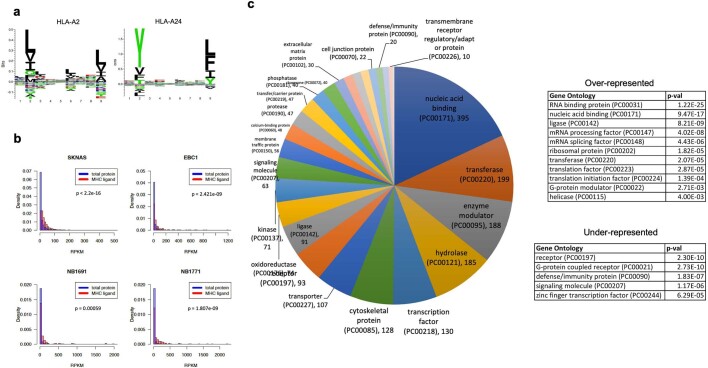
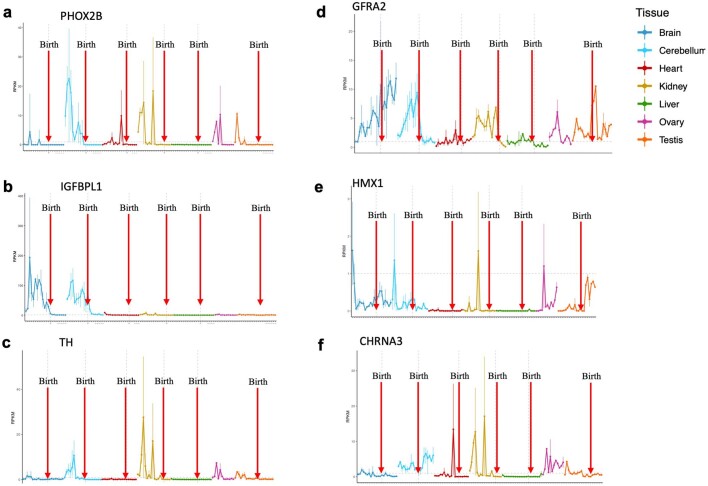
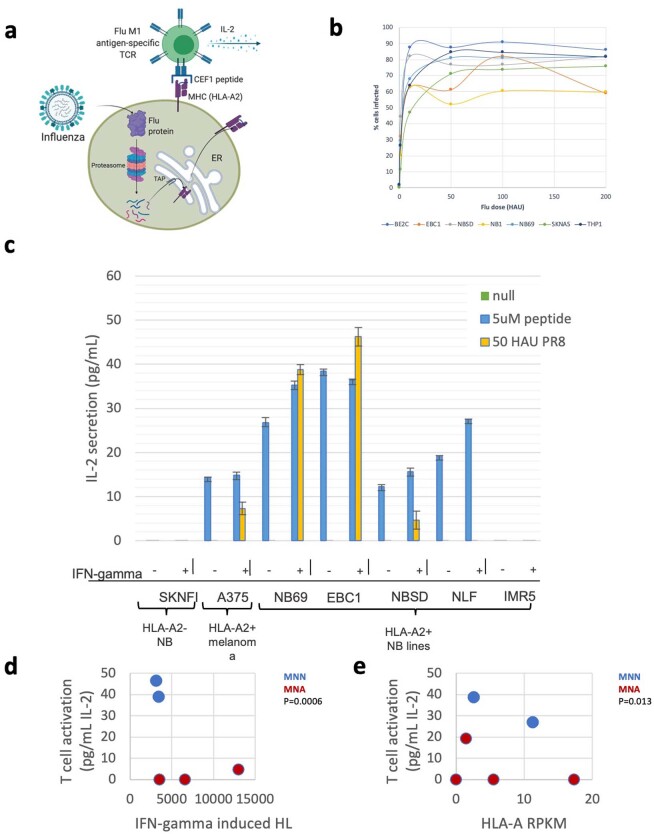
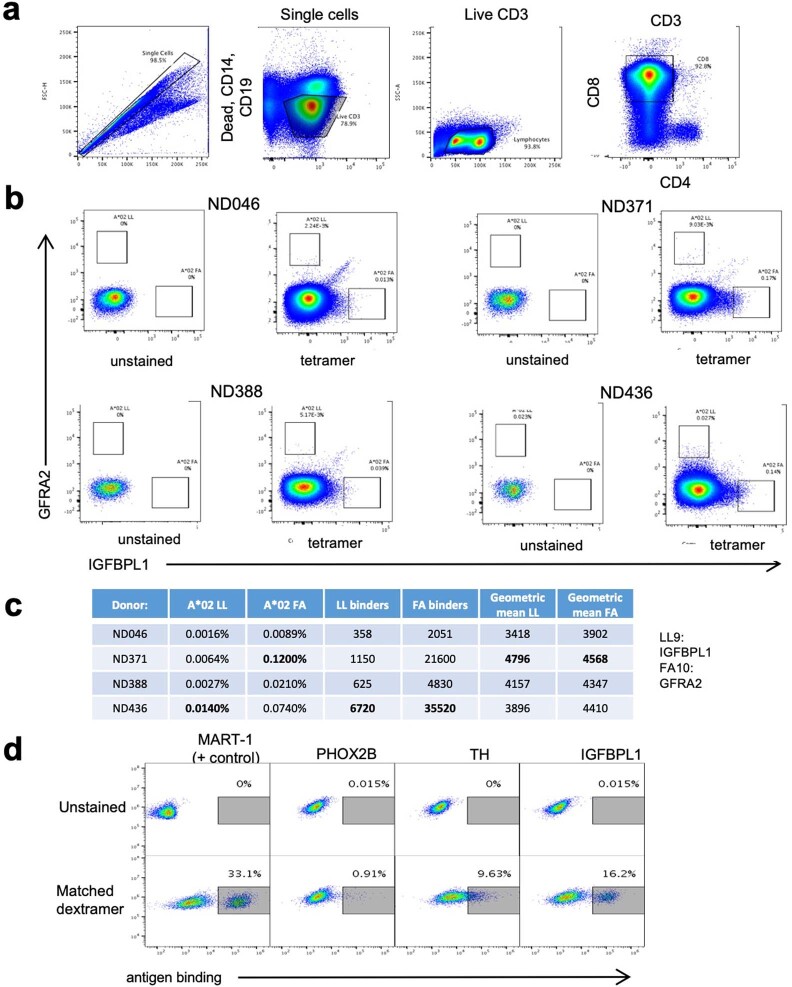
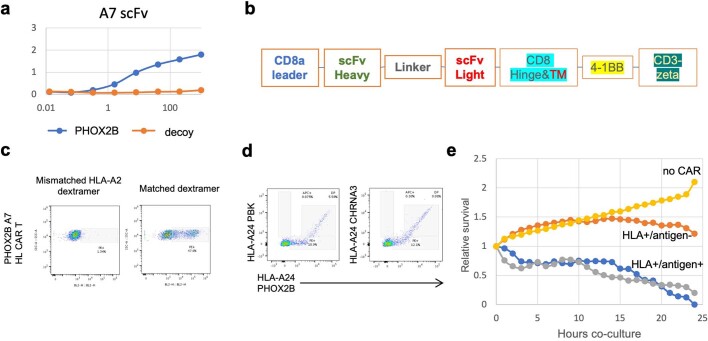
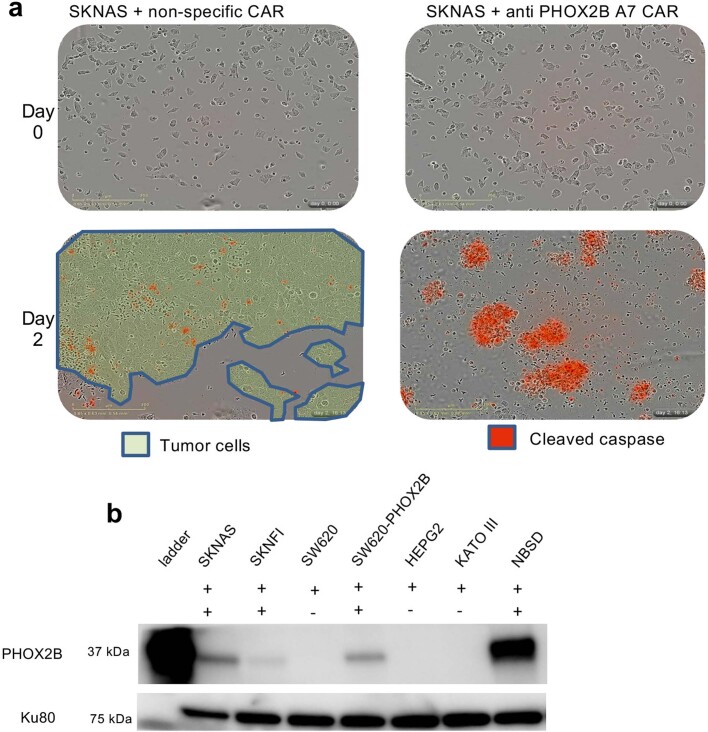
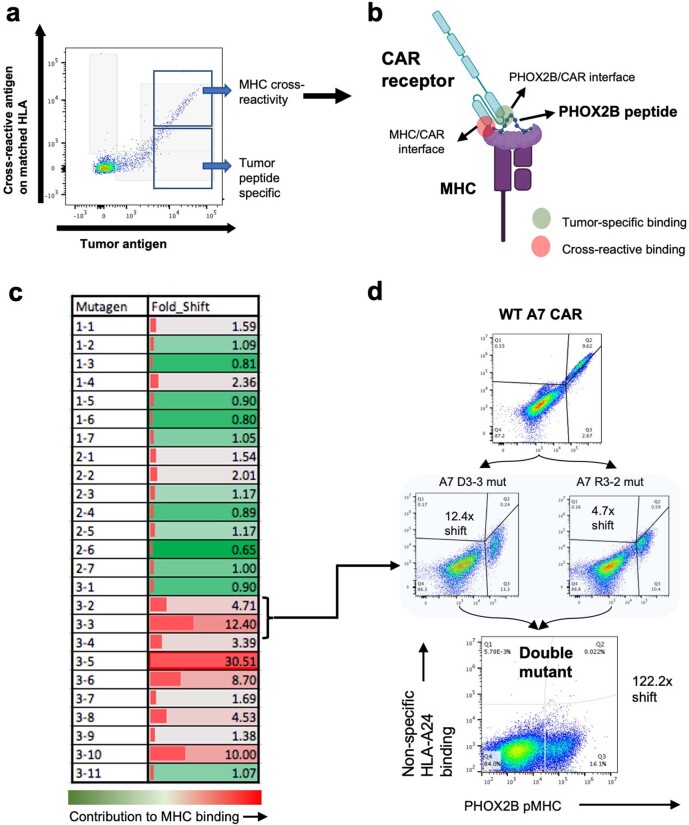
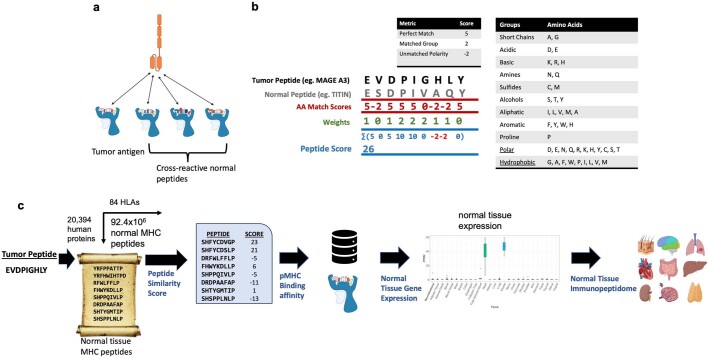
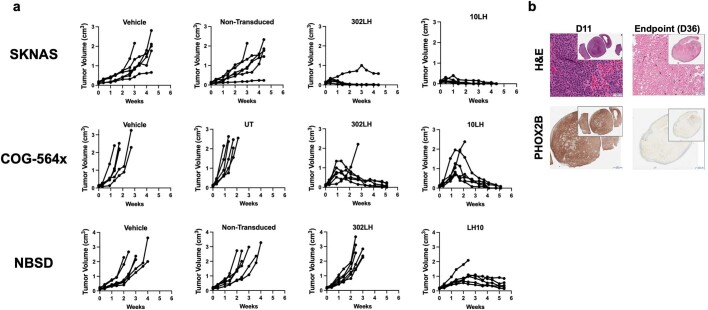
The majority of oncogenic drivers are intracellular proteins, constraining their immunotherapeutic targeting to mutated peptides (neoantigens) presented by individual human leukocyte antigen (HLA) allotypes1. However, most cancers have a modest mutational burden that is insufficient for generating responses using neoantigen-based therapies2,3. Neuroblastoma is a paediatric cancer that harbours few mutations and is instead driven by epigenetically deregulated transcriptional networks4. Here we show that the neuroblastoma immunopeptidome is enriched with peptides derived from proteins essential for tumorigenesis. We focused on targeting the unmutated peptide QYNPIRTTF discovered on HLA-A*24:02, which is derived from the neuroblastoma-dependency gene and master transcriptional regulator PHOX2B. To target QYNPIRTTF, we developed peptide-centric chimeric antigen receptors (PC-CARs) through a counter panning strategy using predicted potentially cross-reactive peptides. We further proposed that PC-CARs can recognize peptides on additional HLA allotypes when presenting a similar overall molecular surface. Informed by our computational modelling results, we show that PHOX2B PC-CARs also recognize QYNPIRTTF presented by HLA-A*23:01, the most common non-A2 allele in people with African ancestry. Finally, we demonstrate potent and specific killing of neuroblastoma cells expressing these HLAs in vitro and complete tumour regression in mice. These data suggest that PC-CARs have the potential to expand the pool of immunotherapeutic targets to include non-immunogenic intracellular oncoproteins and allow targeting through additional HLA allotypes in a clinical setting.
© 2023. The Author(s).
Conflict of interest statement
J.M.M. and M.Y. have equity stake in Tantigen Bio and HuLA Therapeutics. Both companies have interest in commercializing technologies described herein. B.R.K. is an employee of and has equity interests in Myrio Therapeutics. R.P. and N.L.C. are employees of Myrio Therapeutics. J.M.M., M.Y., N.G.S. and B.R.K. are co-inventors on a patent filed regarding the PHOX2B PC-CAR.
Figures
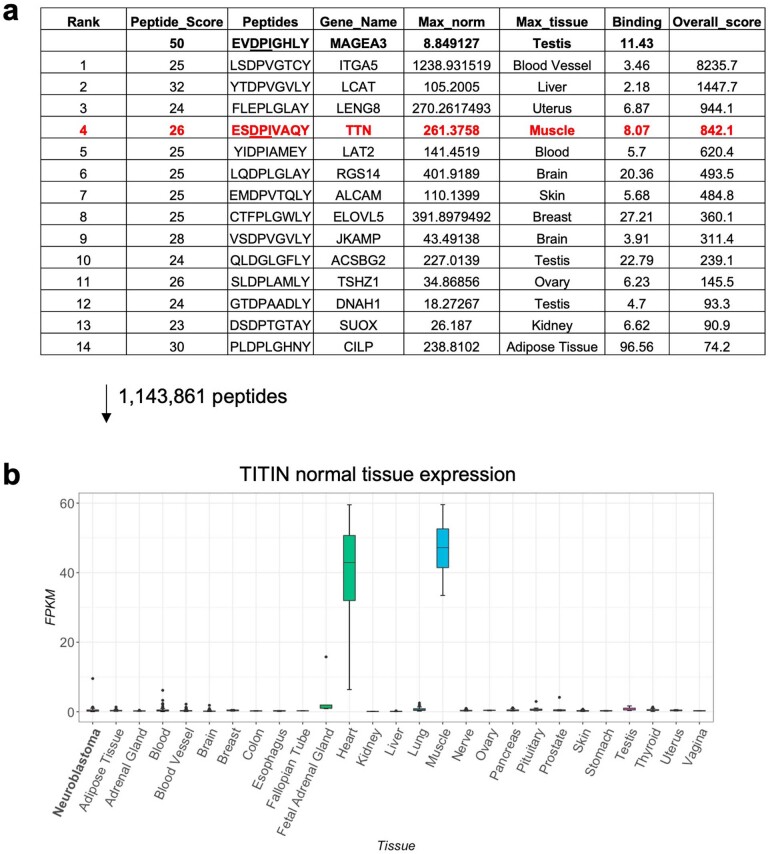
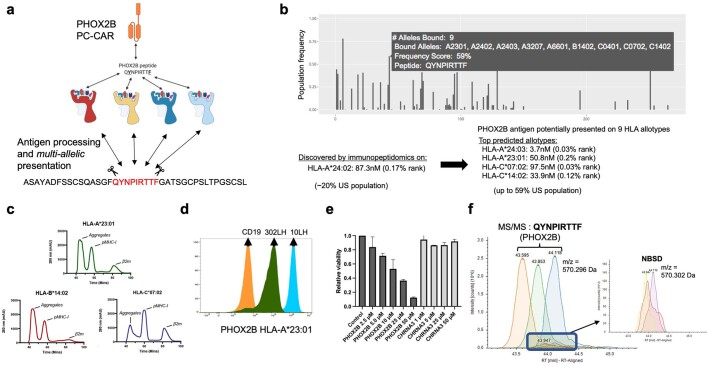
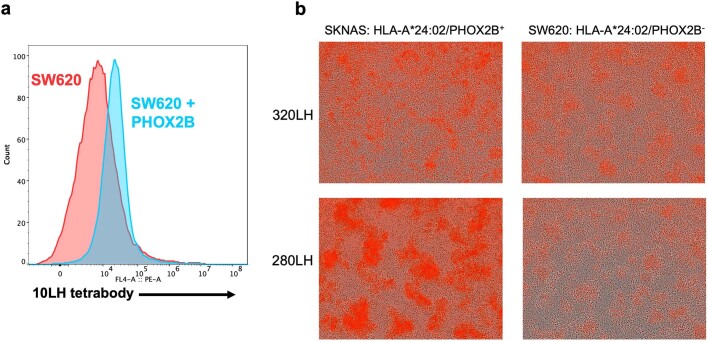
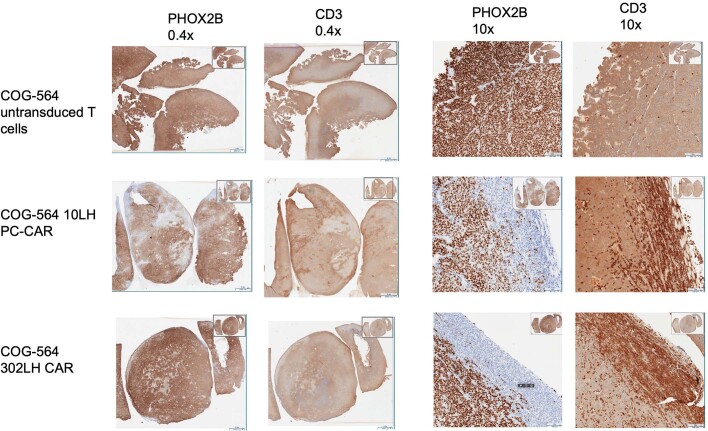
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
