

Autoantibodies against type I IFNs in humans with alternative NF-κB pathway deficiency
- PMID: 37938781
- PMCID: PMC10665196
- DOI: 10.1038/s41586-023-06717-x
Autoantibodies against type I IFNs in humans with alternative NF-κB pathway deficiency
Abstract
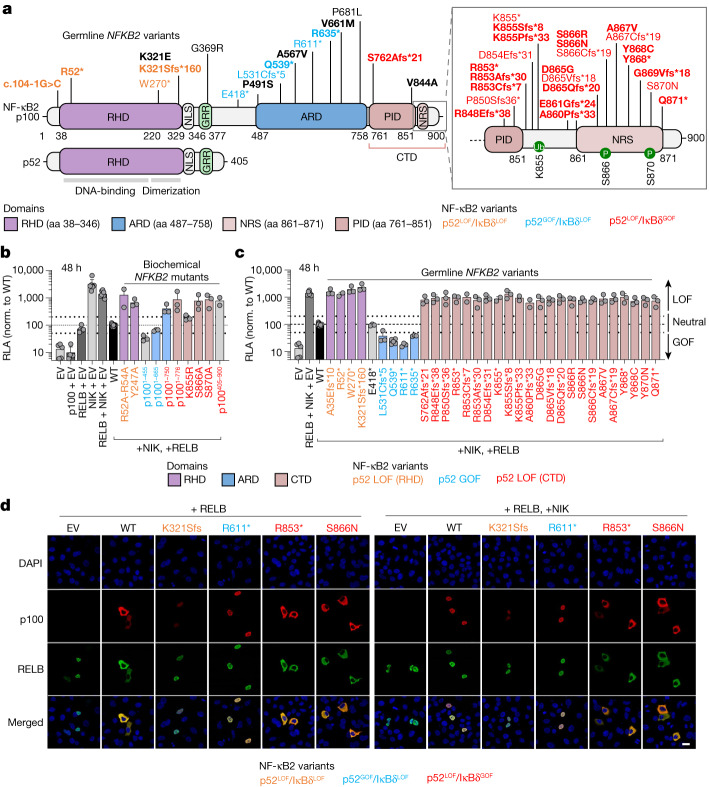
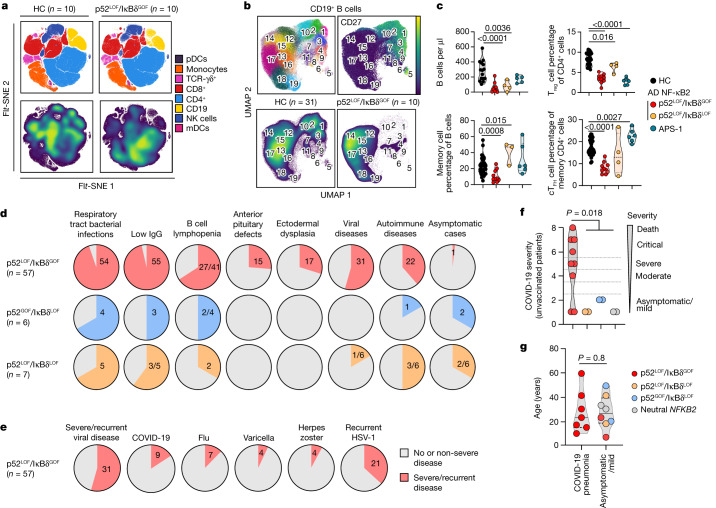
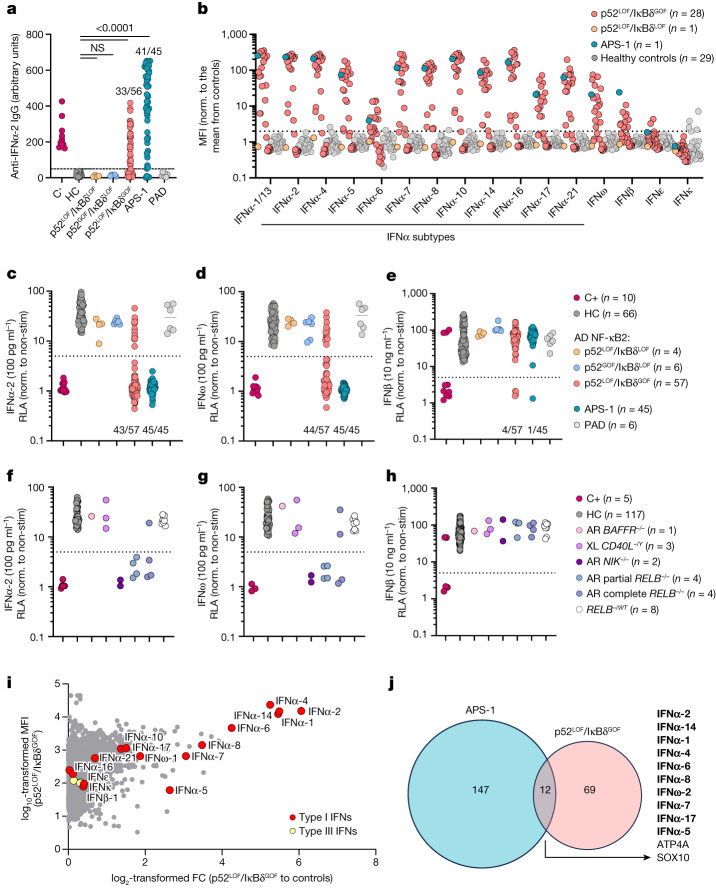
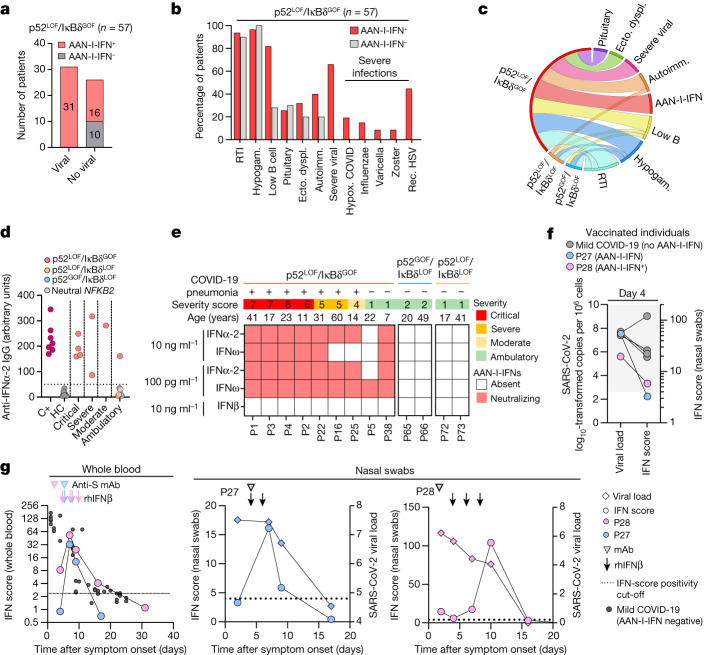
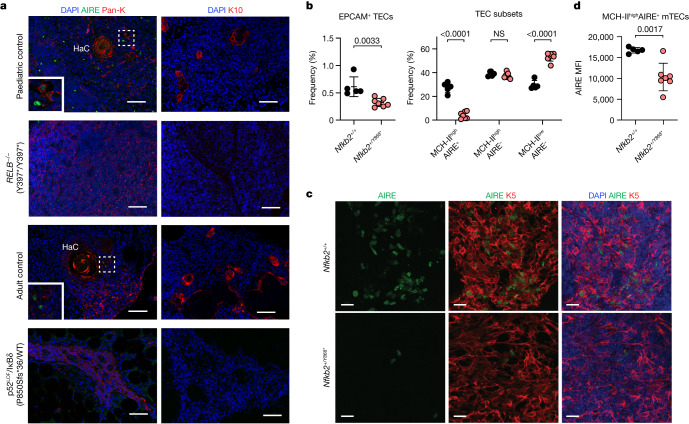
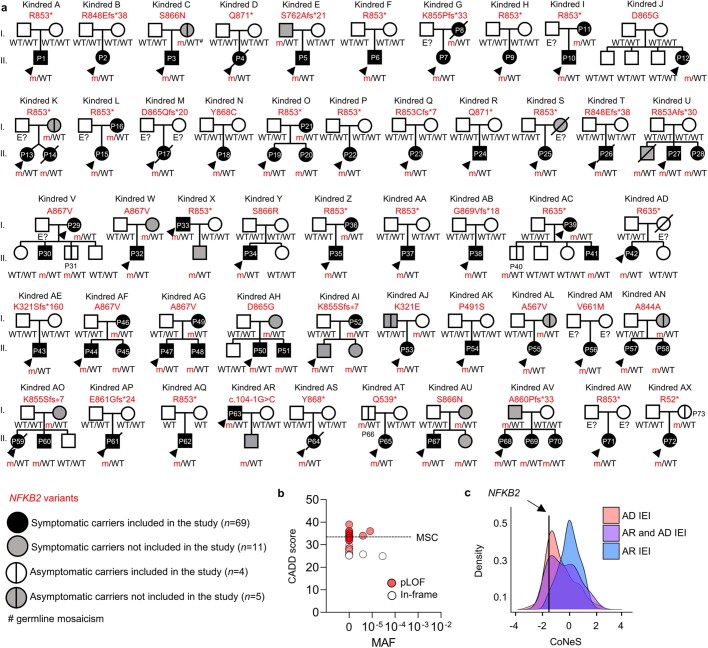
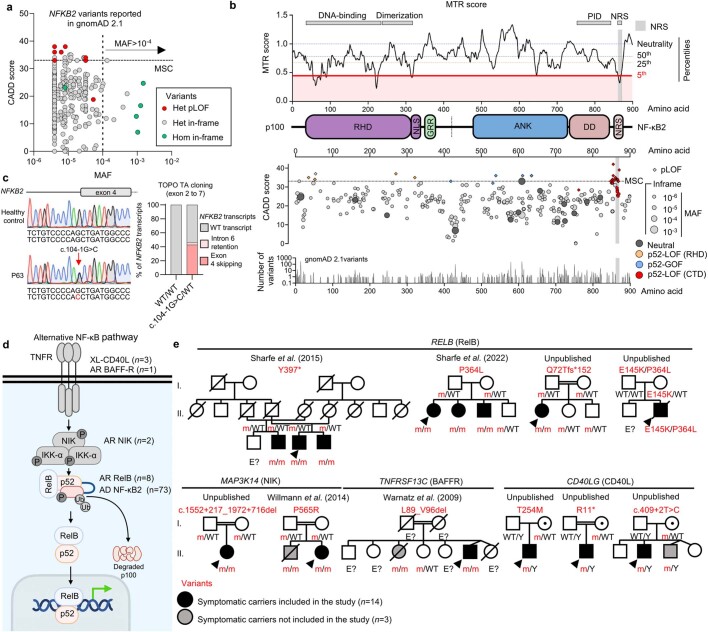
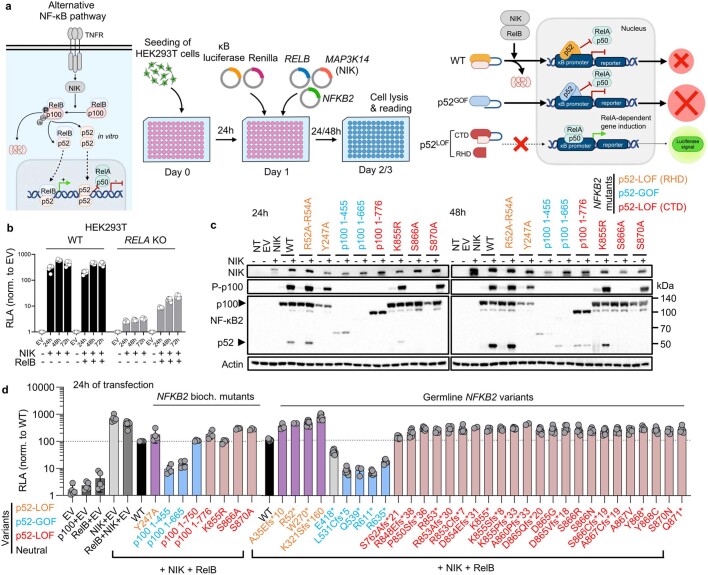
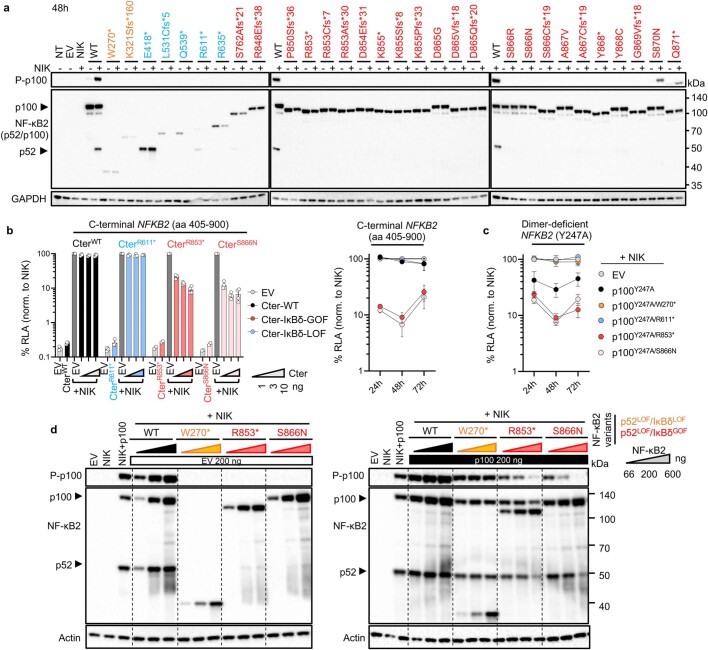
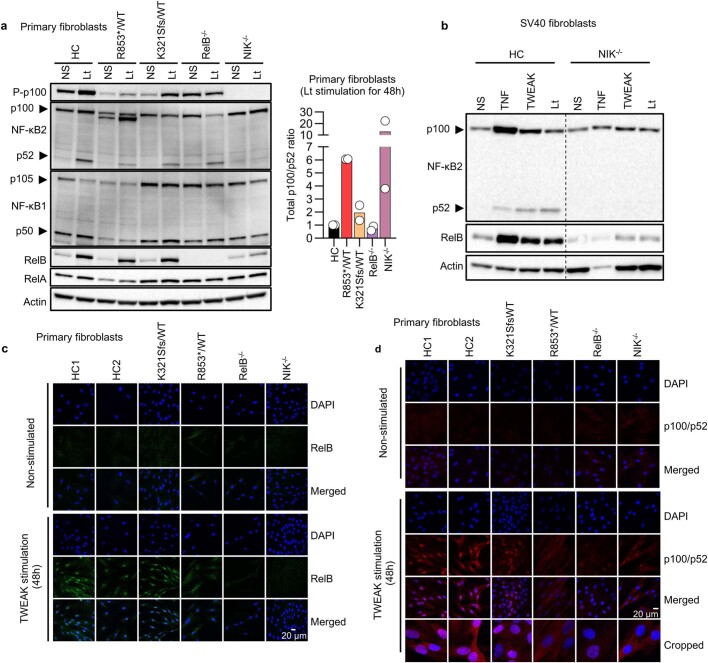
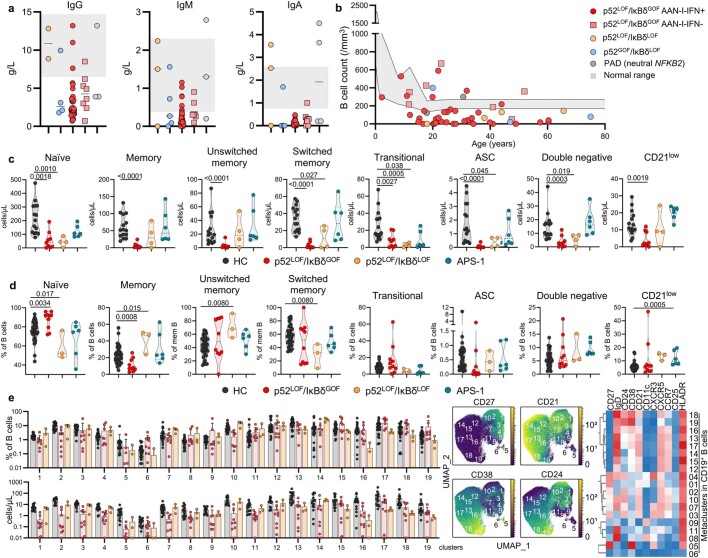
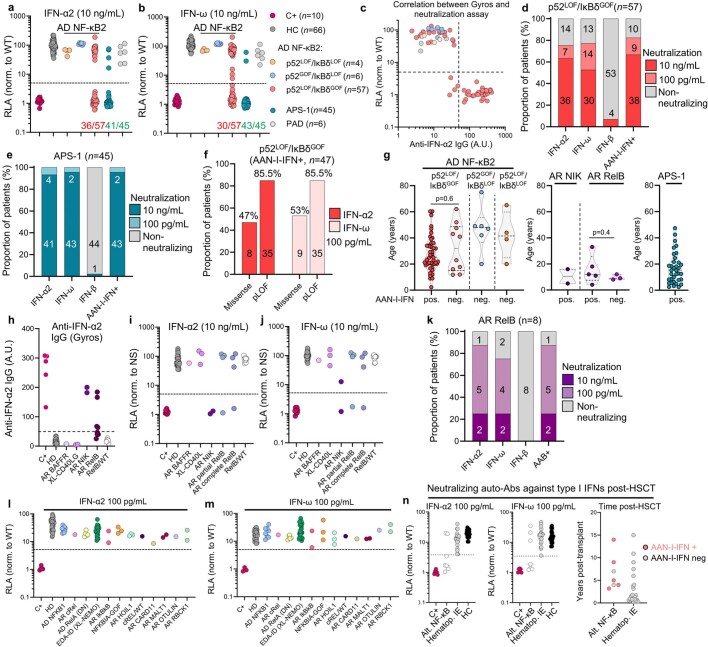
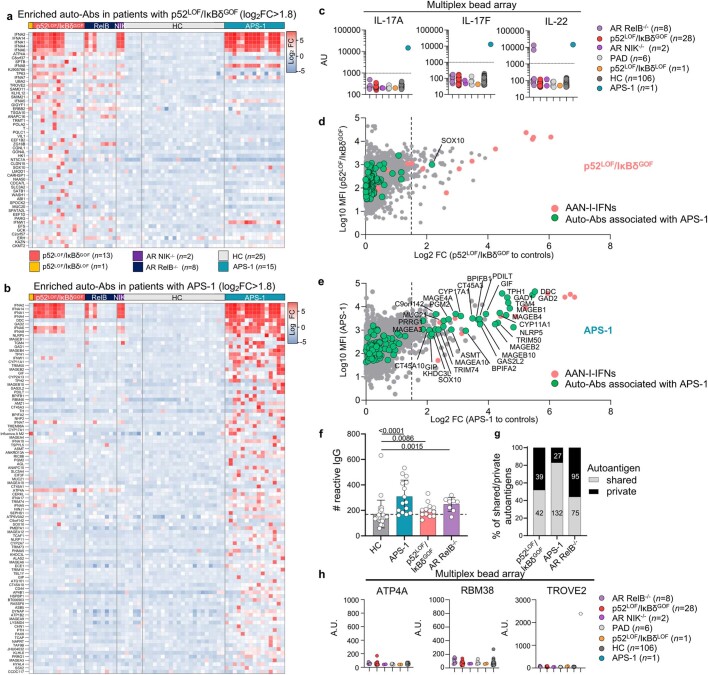
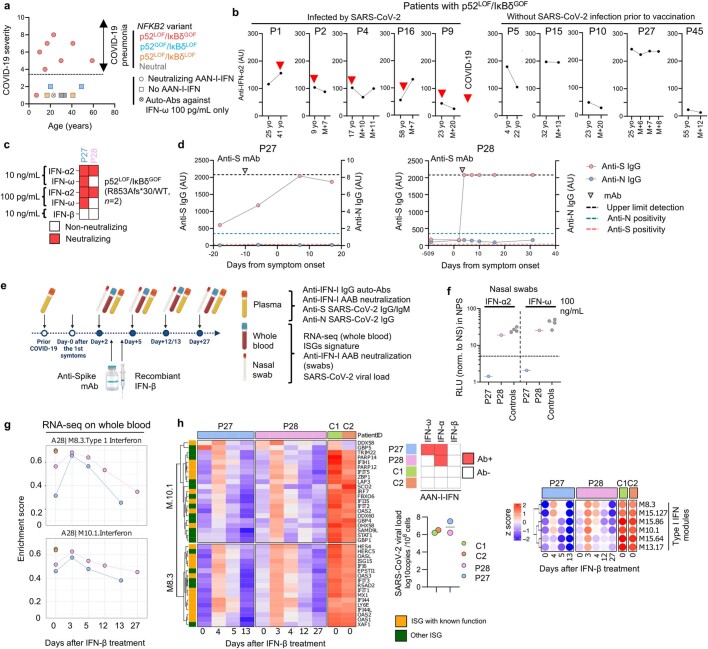
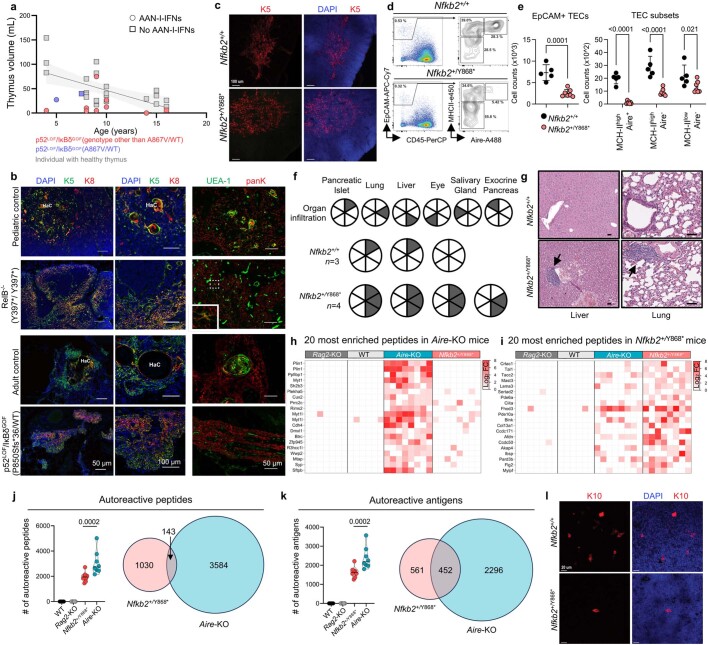
Patients with autoimmune polyendocrinopathy syndrome type 1 (APS-1) caused by autosomal recessive AIRE deficiency produce autoantibodies that neutralize type I interferons (IFNs)1,2, conferring a predisposition to life-threatening COVID-19 pneumonia3. Here we report that patients with autosomal recessive NIK or RELB deficiency, or a specific type of autosomal-dominant NF-κB2 deficiency, also have neutralizing autoantibodies against type I IFNs and are at higher risk of getting life-threatening COVID-19 pneumonia. In patients with autosomal-dominant NF-κB2 deficiency, these autoantibodies are found only in individuals who are heterozygous for variants associated with both transcription (p52 activity) loss of function (LOF) due to impaired p100 processing to generate p52, and regulatory (IκBδ activity) gain of function (GOF) due to the accumulation of unprocessed p100, therefore increasing the inhibitory activity of IκBδ (hereafter, p52LOF/IκBδGOF). By contrast, neutralizing autoantibodies against type I IFNs are not found in individuals who are heterozygous for NFKB2 variants causing haploinsufficiency of p100 and p52 (hereafter, p52LOF/IκBδLOF) or gain-of-function of p52 (hereafter, p52GOF/IκBδLOF). In contrast to patients with APS-1, patients with disorders of NIK, RELB or NF-κB2 have very few tissue-specific autoantibodies. However, their thymuses have an abnormal structure, with few AIRE-expressing medullary thymic epithelial cells. Human inborn errors of the alternative NF-κB pathway impair the development of AIRE-expressing medullary thymic epithelial cells, thereby underlying the production of autoantibodies against type I IFNs and predisposition to viral diseases.
© 2023. The Author(s).
Conflict of interest statement
A. Guichard is an employee of Biomerieux. J.-L. Casanova is listed as an inventor on patent application PCT/US2021/042741, filed on the 22 July 2021 and submitted by The Rockefeller University, that covers the diagnosis of susceptibility to, and treatment of, viral disease and viral vaccines, including COVID-19 and vaccine-associated diseases. The other authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
