

Targeting ribosome biogenesis reinforces ERK-dependent senescence in pancreatic cancer
- PMID: 37942963
- PMCID: PMC10732607
- DOI: 10.1080/15384101.2023.2278945
Targeting ribosome biogenesis reinforces ERK-dependent senescence in pancreatic cancer
Abstract
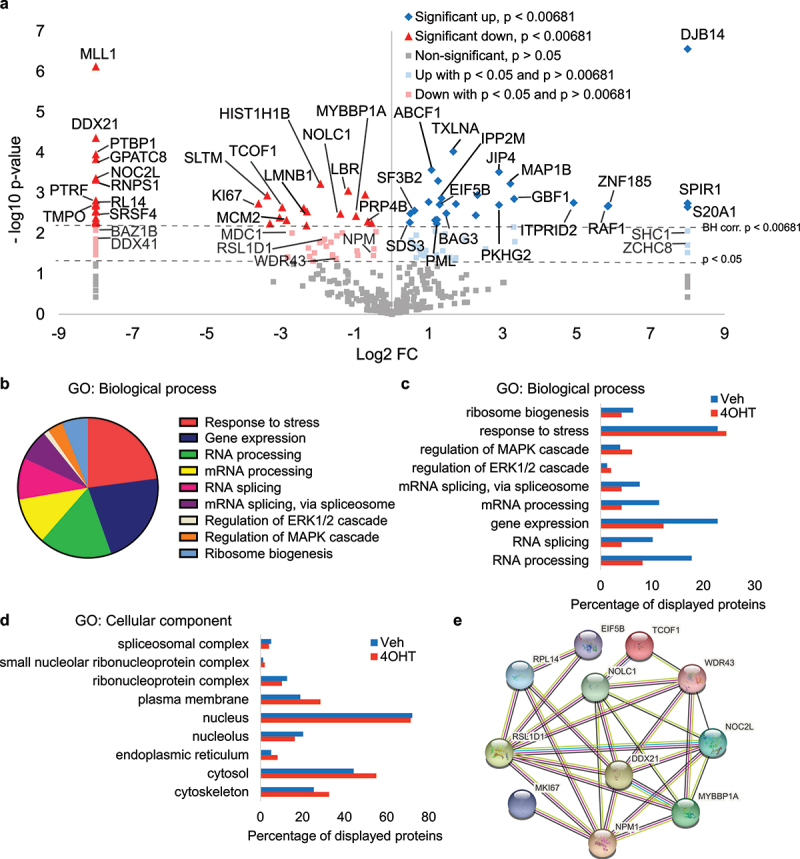
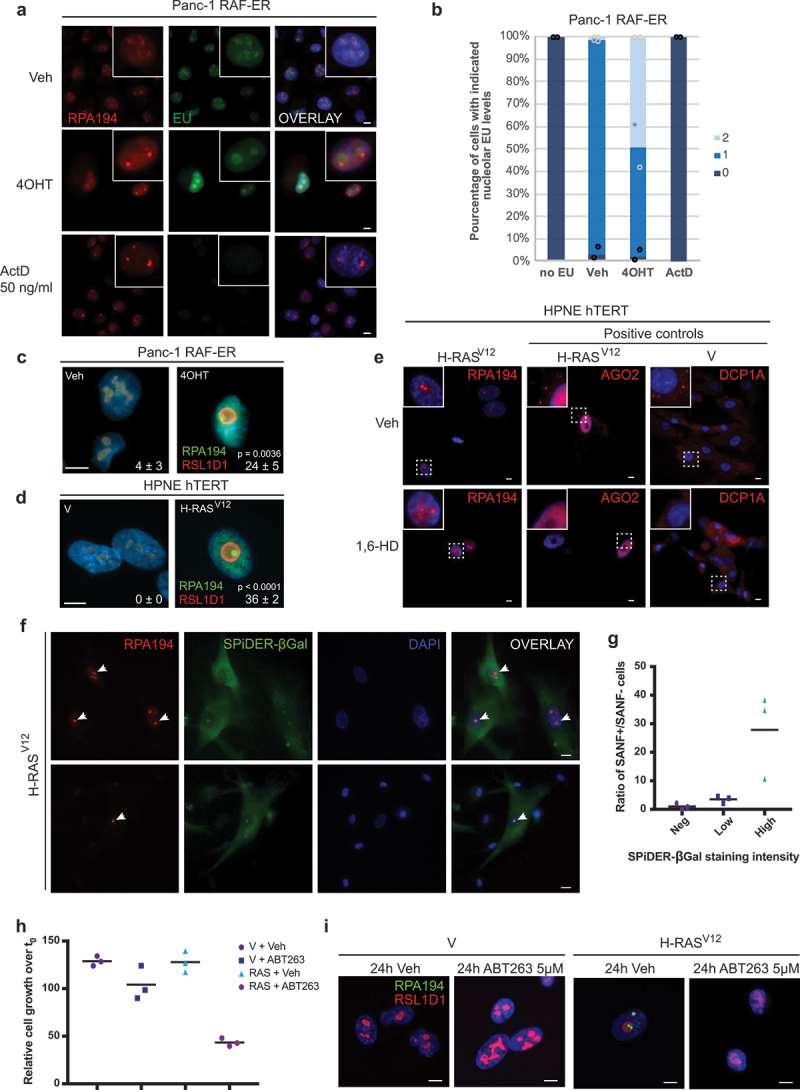
Pancreatic adenocarcinomas (PDAC) often possess mutations in K-Ras that stimulate the ERK pathway. Aberrantly high ERK activation triggers oncogene-induced senescence, which halts tumor progression. Here we report that low-grade pancreatic intraepithelial neoplasia displays very high levels of phospho-ERK consistent with a senescence response. However, advanced lesions that have circumvented the senescence barrier exhibit lower phospho-ERK levels. Restoring ERK hyperactivation in PDAC using activated RAF leads to ERK-dependent growth arrest with senescence biomarkers. ERK-dependent senescence in PDAC was characterized by a nucleolar stress response including a selective depletion of nucleolar phosphoproteins and intranucleolar foci containing RNA polymerase I designated as senescence-associated nucleolar foci (SANF). Accordingly, combining ribosome biogenesis inhibitors with ERK hyperactivation reinforced the senescence response in PDAC cells. Notably, comparable mechanisms were observed upon treatment with the platinum-based chemotherapy regimen FOLFIRINOX, currently a first-line treatment option for PDAC. We thus suggest that drugs targeting ribosome biogenesis can improve the senescence anticancer response in pancreatic cancer.
Keywords: MAP kinase; folfirinox; nucleolus; pancreatic cancer; ribosome biogenesis; therapy-induced senescence.
Conflict of interest statement
No potential conflict of interest was reported by the author(s).
Figures





References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous