

Biallelic Cys141Tyr variant of SEL1L is associated with neurodevelopmental disorders, agammaglobulinemia, and premature death
- PMID: 37943617
- PMCID: PMC10786703
- DOI: 10.1172/JCI170882
Biallelic Cys141Tyr variant of SEL1L is associated with neurodevelopmental disorders, agammaglobulinemia, and premature death
Abstract
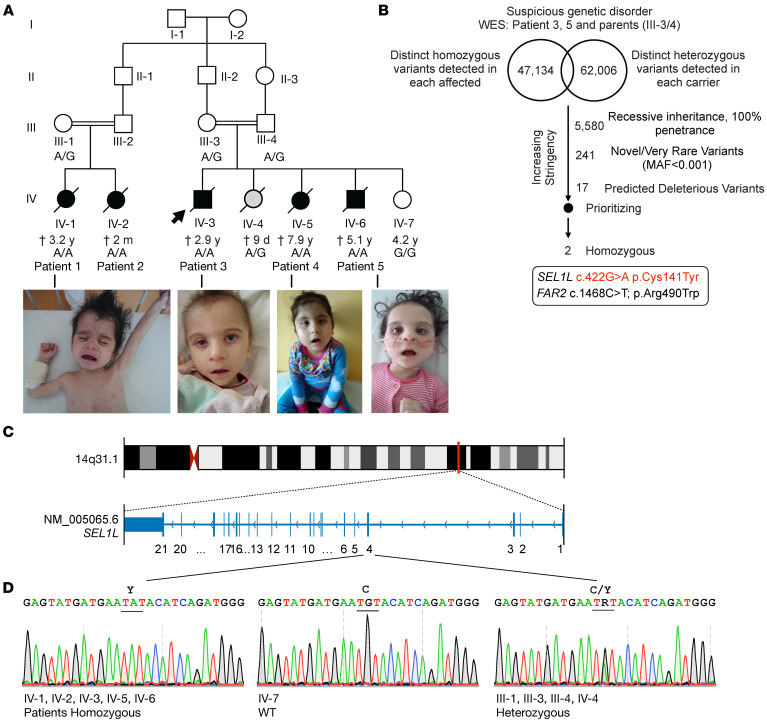
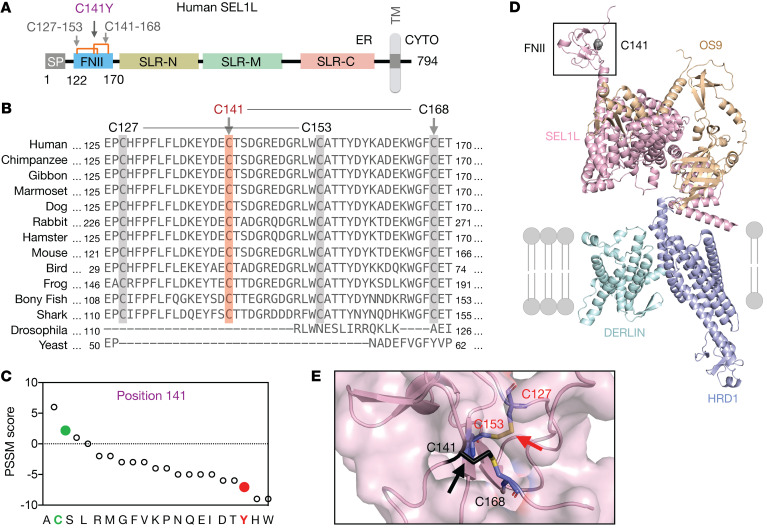
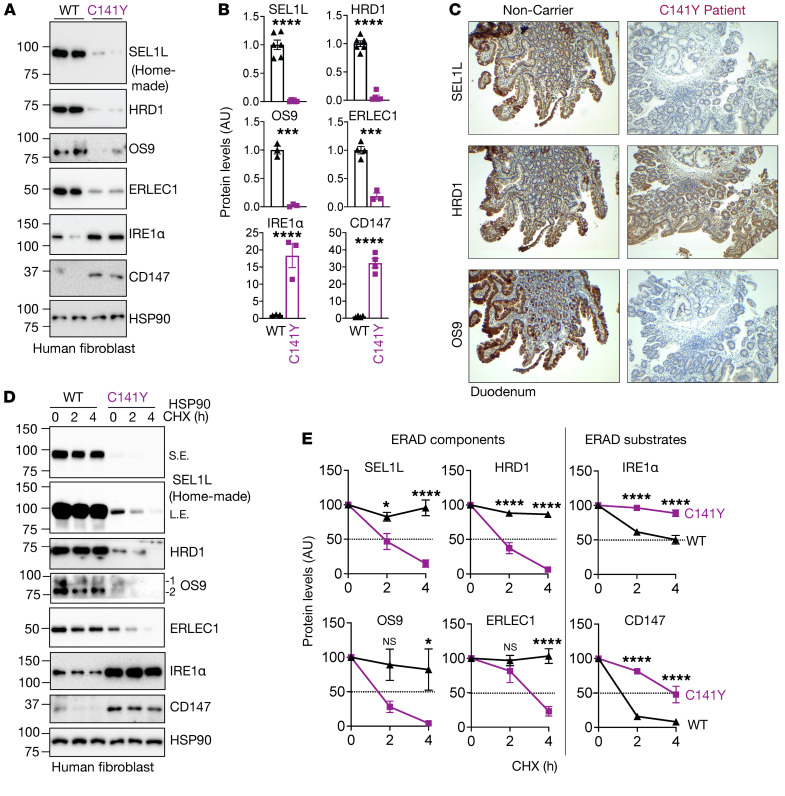
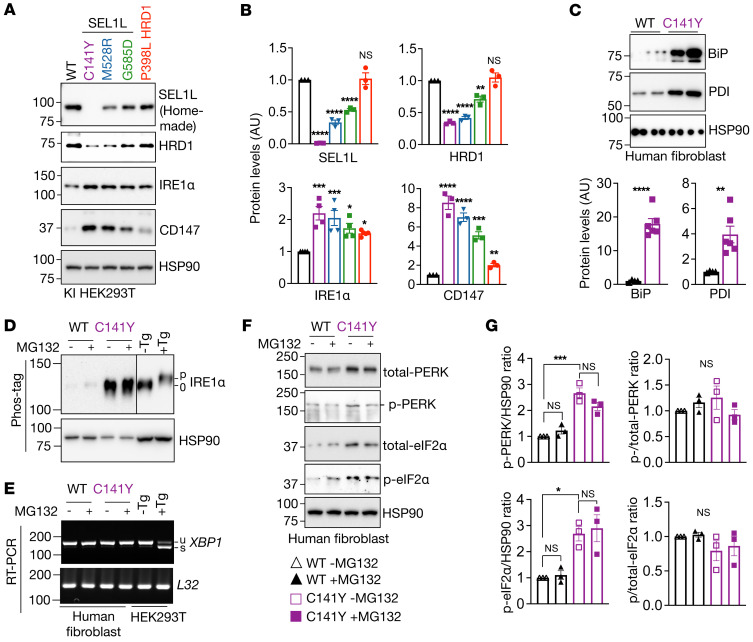
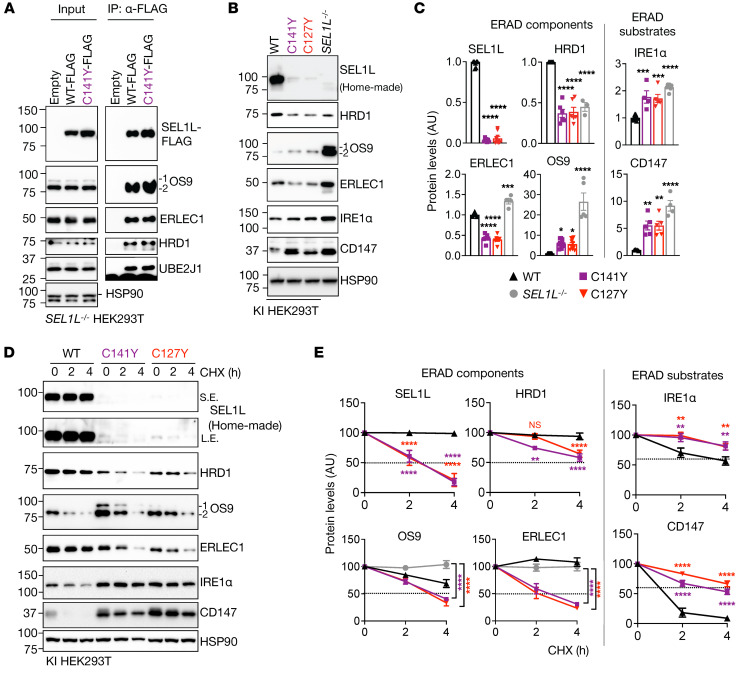
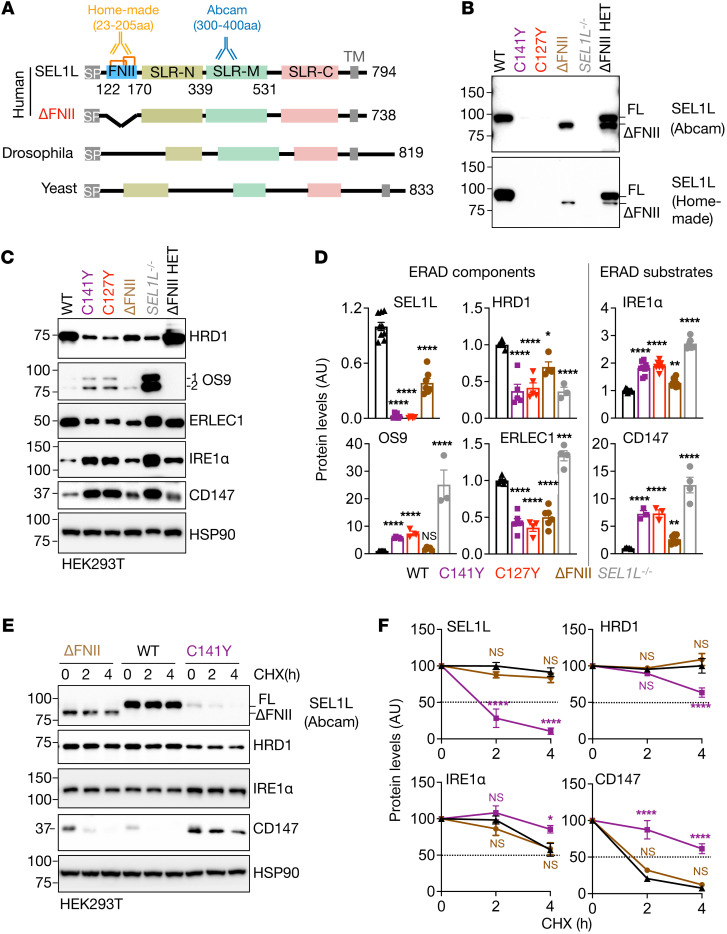
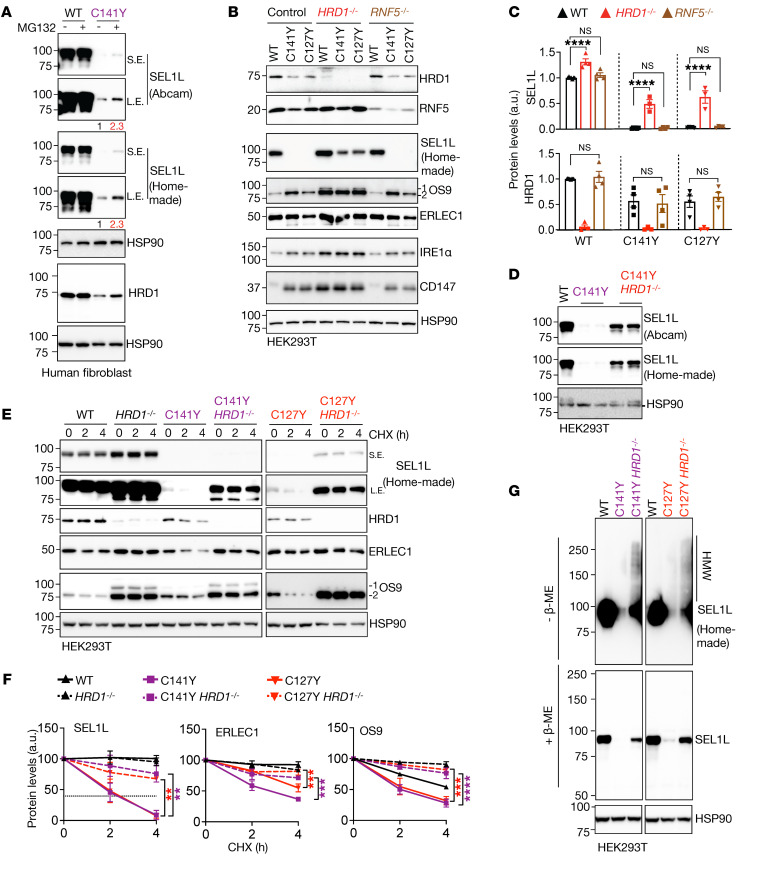
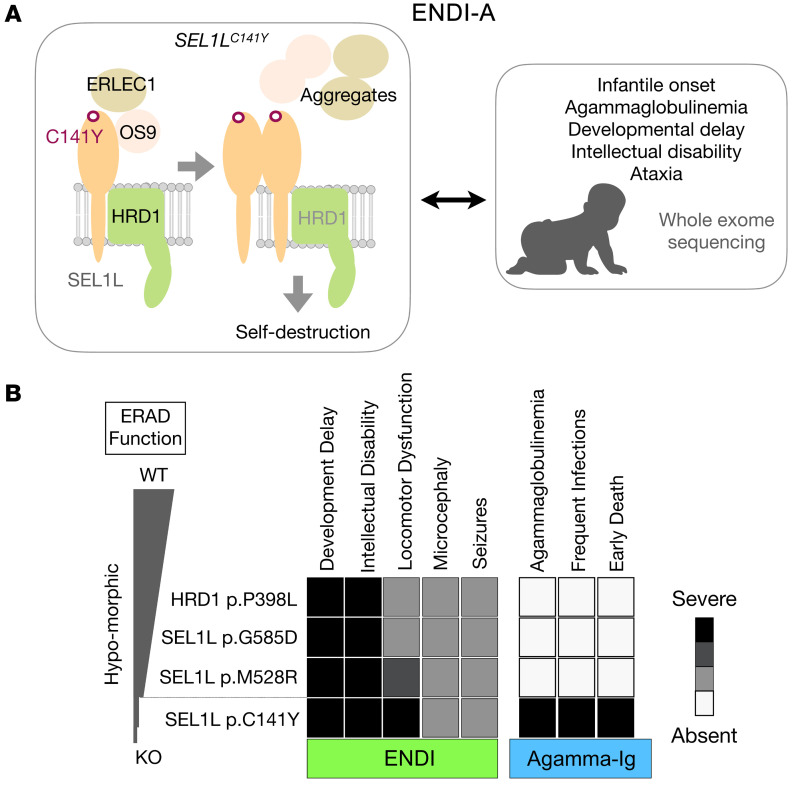
Suppressor of lin-12-like-HMG-CoA reductase degradation 1 (SEL1L-HRD1) ER-associated degradation (ERAD) plays a critical role in many physiological processes in mice, including immunity, water homeostasis, and energy metabolism; however, its relevance and importance in humans remain unclear, as no disease variant has been identified. Here, we report a biallelic SEL1L variant (p. Cys141Tyr) in 5 patients from a consanguineous Slovakian family. These patients presented with not only ERAD-associated neurodevelopmental disorders with onset in infancy (ENDI) syndromes, but infantile-onset agammaglobulinemia with no mature B cells, resulting in frequent infections and early death. This variant disrupted the formation of a disulfide bond in the luminal fibronectin II domain of SEL1L, largely abolishing the function of the SEL1L-HRD1 ERAD complex in part via proteasomal-mediated self destruction by HRD1. This study reports a disease entity termed ENDI-agammaglobulinemia (ENDI-A) syndrome and establishes an inverse correlation between SEL1L-HRD1 ERAD functionality and disease severity in humans.
Keywords: Adaptive immunity; Cell Biology; Genetic diseases; Protein misfolding.
Figures
Comment in
- Hypomorphic human SEL1L and HRD1 variants uncouple multilayered ER-associated degradation machinery
Similar articles
-
SEL1L-HRD1-mediated ERAD in mammals.Nat Cell Biol. 2025 Jul;27(7):1063-1073. doi: 10.1038/s41556-025-01690-1. Epub 2025 Jun 25. Nat Cell Biol. 2025. PMID: 40562846 Free PMC article. Review.
-
Hypomorphic variants of SEL1L-HRD1 ER-associated degradation are associated with neurodevelopmental disorders.J Clin Invest. 2024 Jan 16;134(2):e170054. doi: 10.1172/JCI170054. J Clin Invest. 2024. PMID: 37943610 Free PMC article.
-
Functional rescue of a fatal ERAD mutation via alternative splicing.bioRxiv [Preprint]. 2025 Jun 15:2025.06.13.659581. doi: 10.1101/2025.06.13.659581. bioRxiv. 2025. PMID: 40661444 Free PMC article. Preprint.
-
SEL1L-HRD1 interaction is required to form a functional HRD1 ERAD complex.Nat Commun. 2024 Feb 16;15(1):1440. doi: 10.1038/s41467-024-45633-0. Nat Commun. 2024. PMID: 38365914 Free PMC article.
-
Endoplasmic reticulum (ER) protein degradation by ER-associated degradation and ER-phagy.Trends Cell Biol. 2025 Jul;35(7):576-591. doi: 10.1016/j.tcb.2025.01.002. Epub 2025 Feb 4. Trends Cell Biol. 2025. PMID: 39909774 Review.
Cited by
-
SEL1L-HRD1-mediated ERAD in mammals.Nat Cell Biol. 2025 Jul;27(7):1063-1073. doi: 10.1038/s41556-025-01690-1. Epub 2025 Jun 25. Nat Cell Biol. 2025. PMID: 40562846 Free PMC article. Review.
-
Genome-wide screens identify SEL1L as an intracellular rheostat controlling collagen turnover.Nat Commun. 2024 Feb 20;15(1):1531. doi: 10.1038/s41467-024-45817-8. Nat Commun. 2024. PMID: 38378719 Free PMC article.
-
Proteomic screens of SEL1L-HRD1 ER-associated degradation substrates reveal its role in glycosylphosphatidylinositol-anchored protein biogenesis.Nat Commun. 2024 Jan 22;15(1):659. doi: 10.1038/s41467-024-44948-2. Nat Commun. 2024. PMID: 38253565 Free PMC article.
-
Hypomorphic human SEL1L and HRD1 variants uncouple multilayered ER-associated degradation machinery.J Clin Invest. 2024 Jan 16;134(2):e175448. doi: 10.1172/JCI175448. J Clin Invest. 2024. PMID: 38226624 Free PMC article.
-
Neuronal SEL1L-HRD1 ERAD regulates one-carbon metabolism and is essential for motor function and survival.bioRxiv [Preprint]. 2025 Jun 18:2025.06.16.659938. doi: 10.1101/2025.06.16.659938. bioRxiv. 2025. PMID: 40667142 Free PMC article. Preprint.
