

How Robust Is the Ligand Binding Transition State?
- PMID: 37943667
- PMCID: PMC11059145
- DOI: 10.1021/jacs.3c08940
How Robust Is the Ligand Binding Transition State?
Abstract
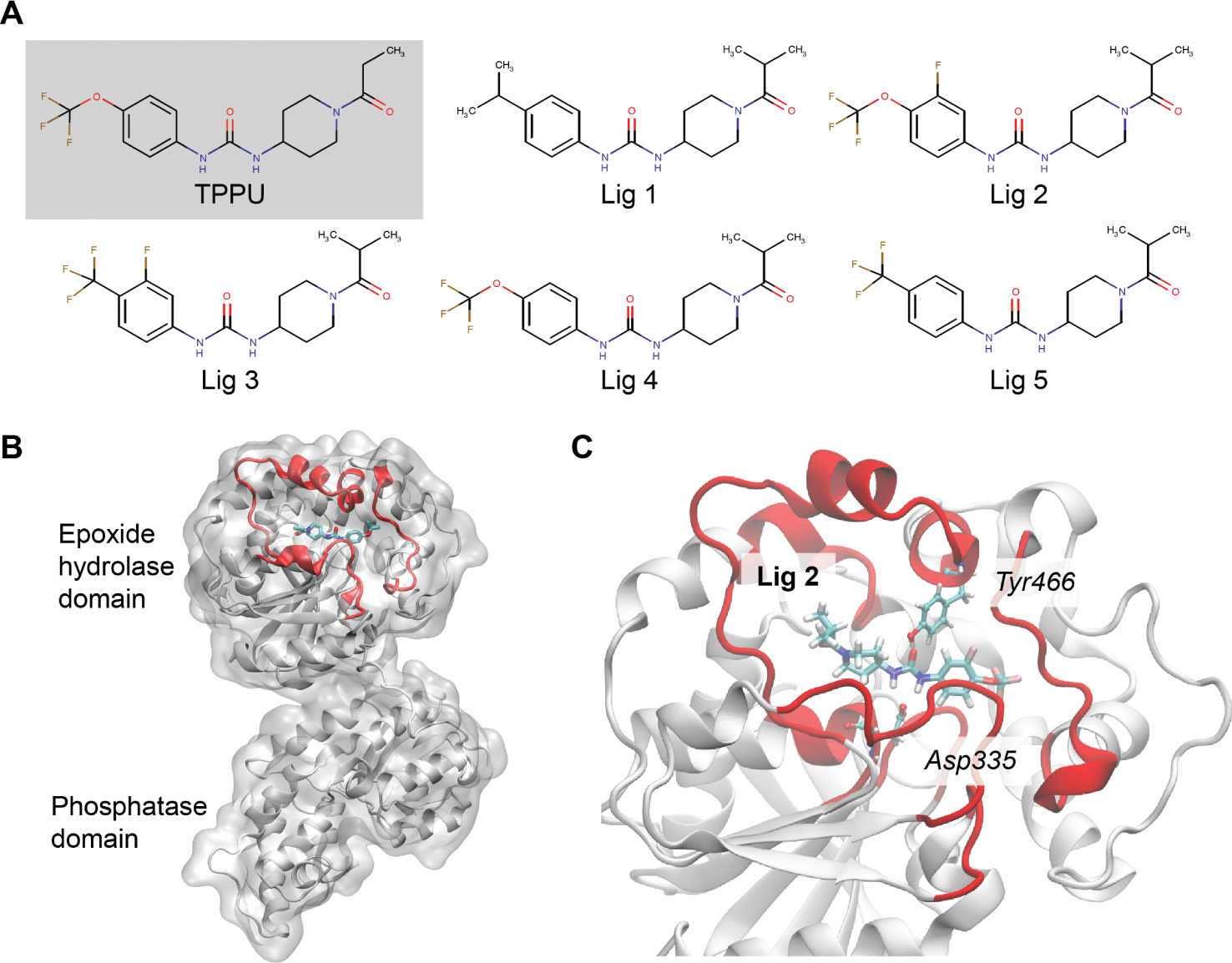
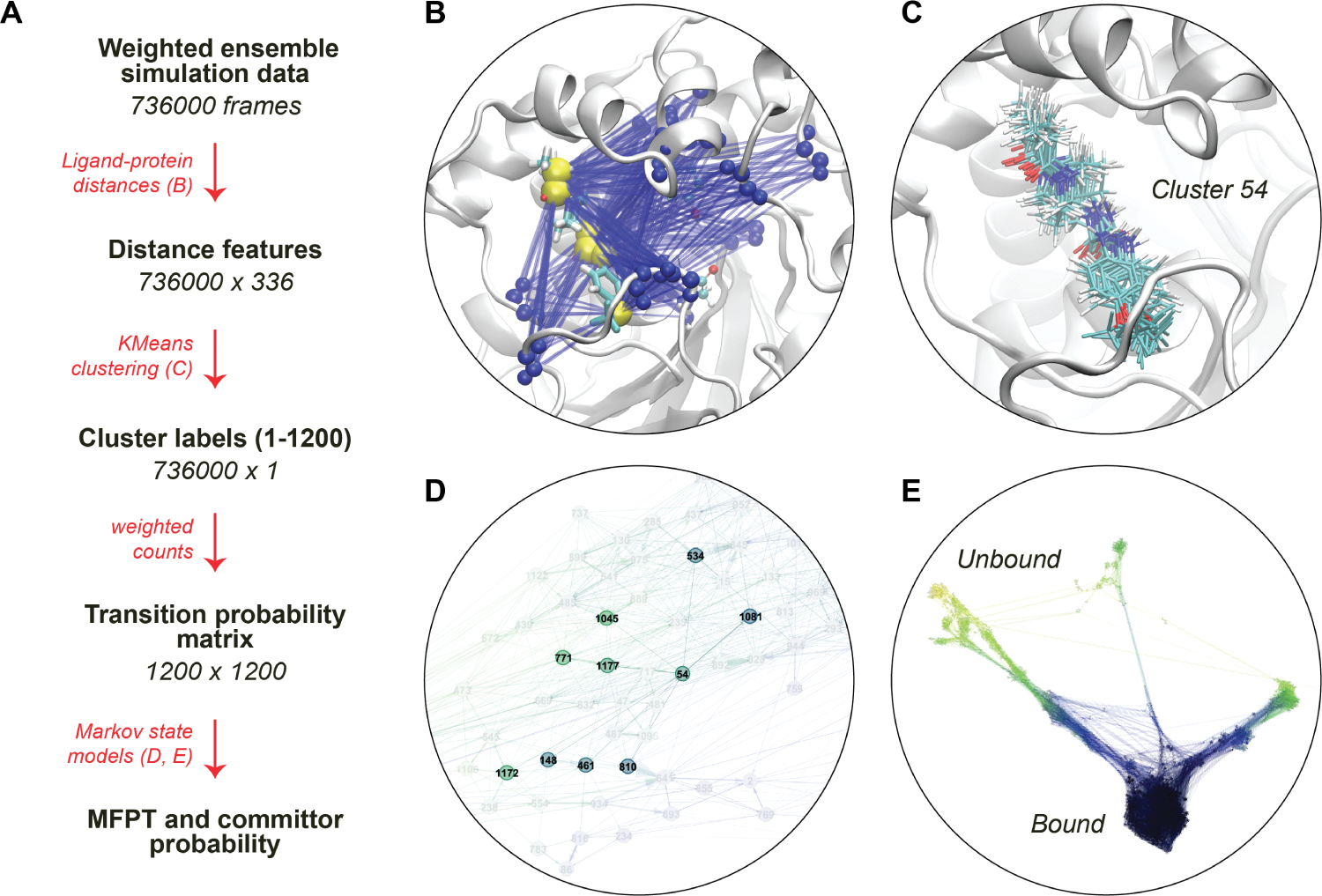
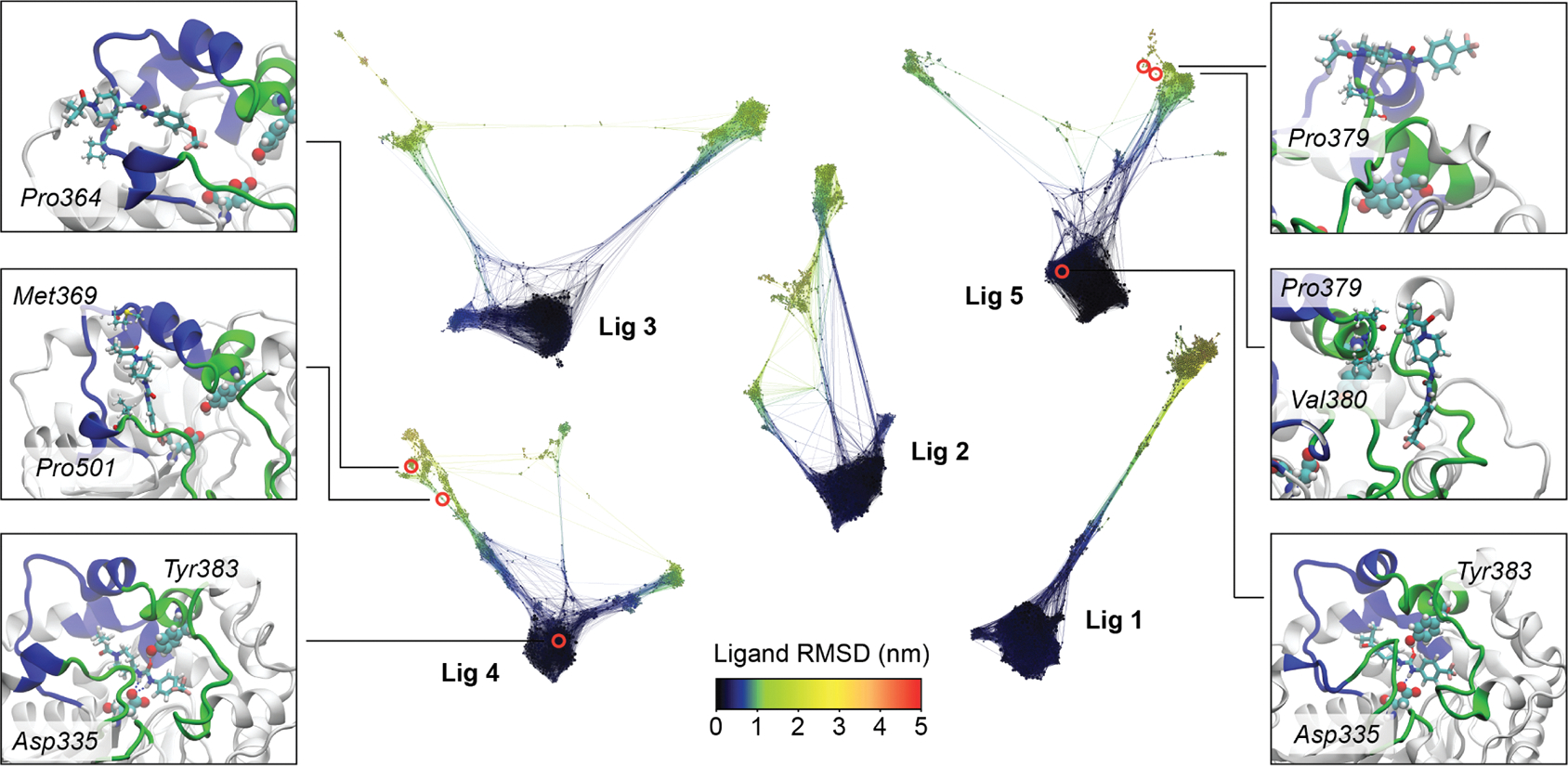
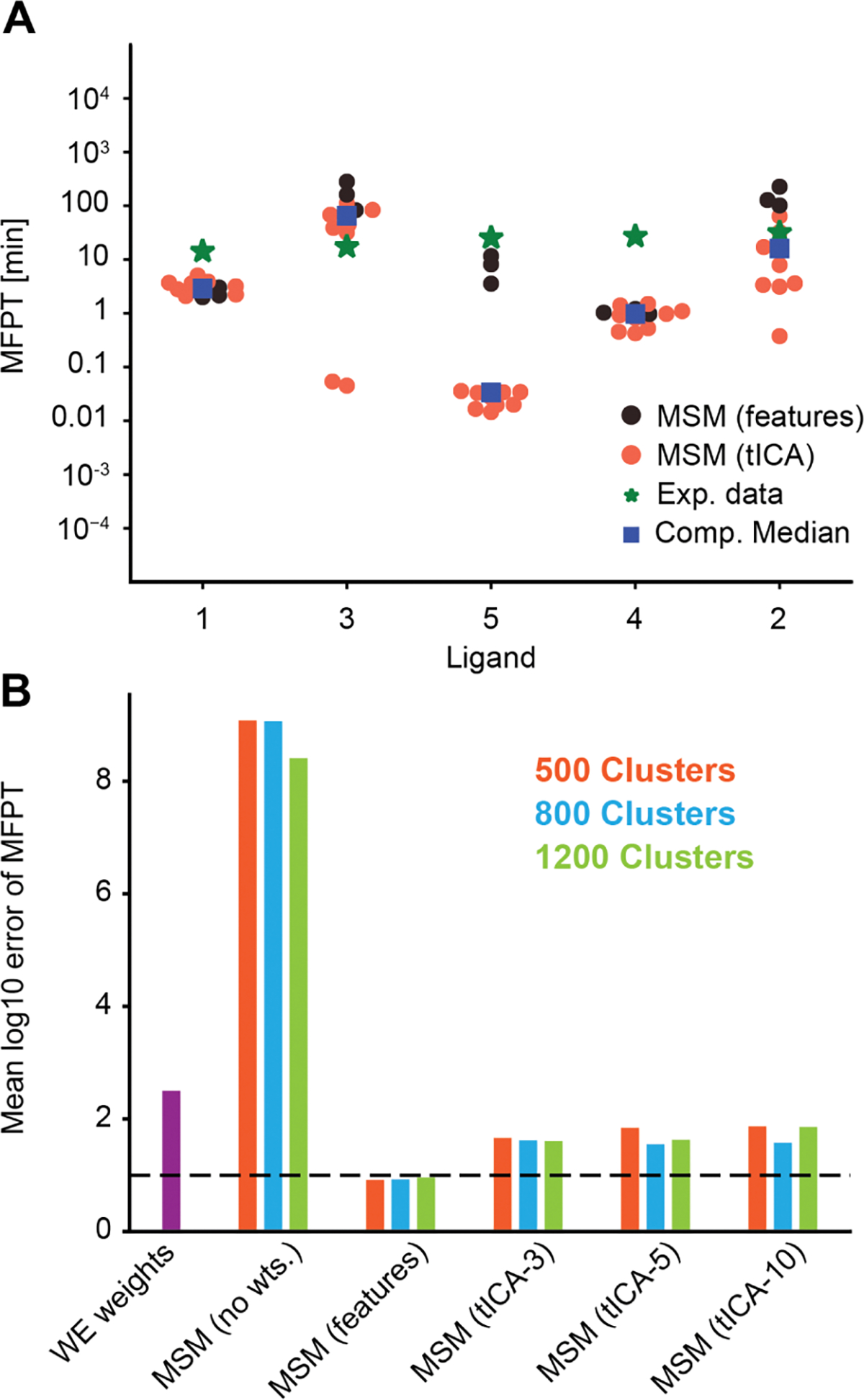
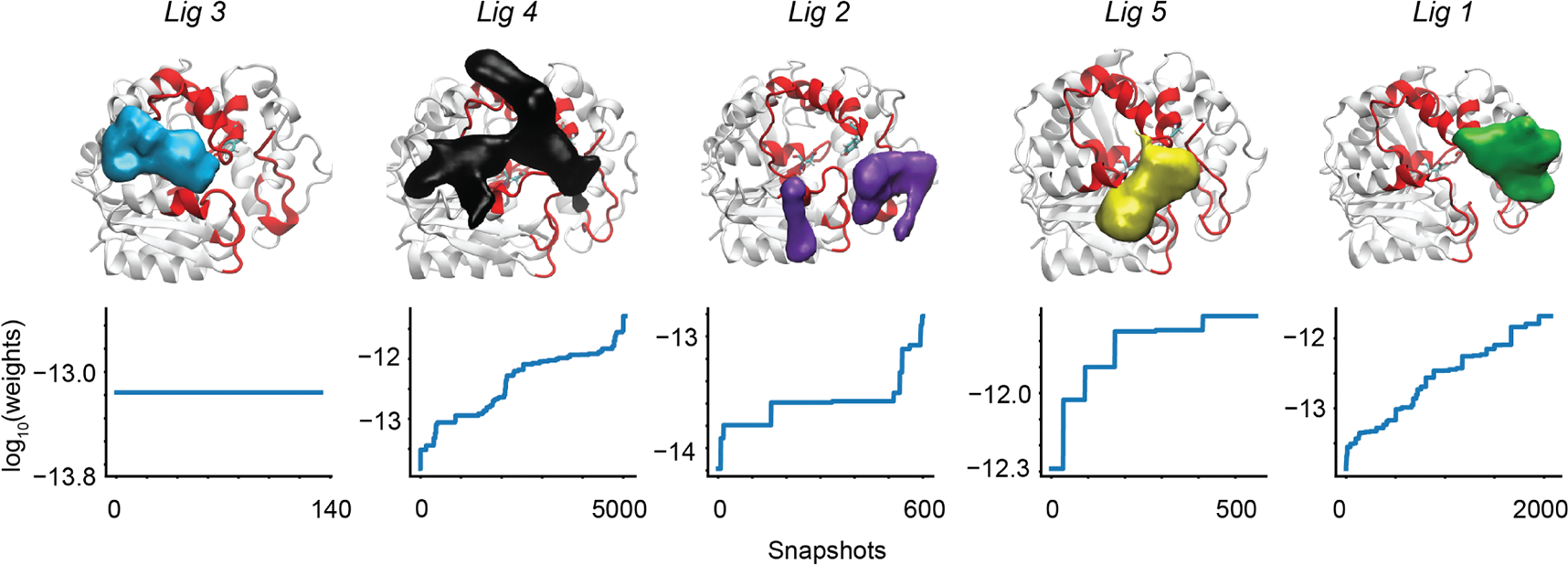
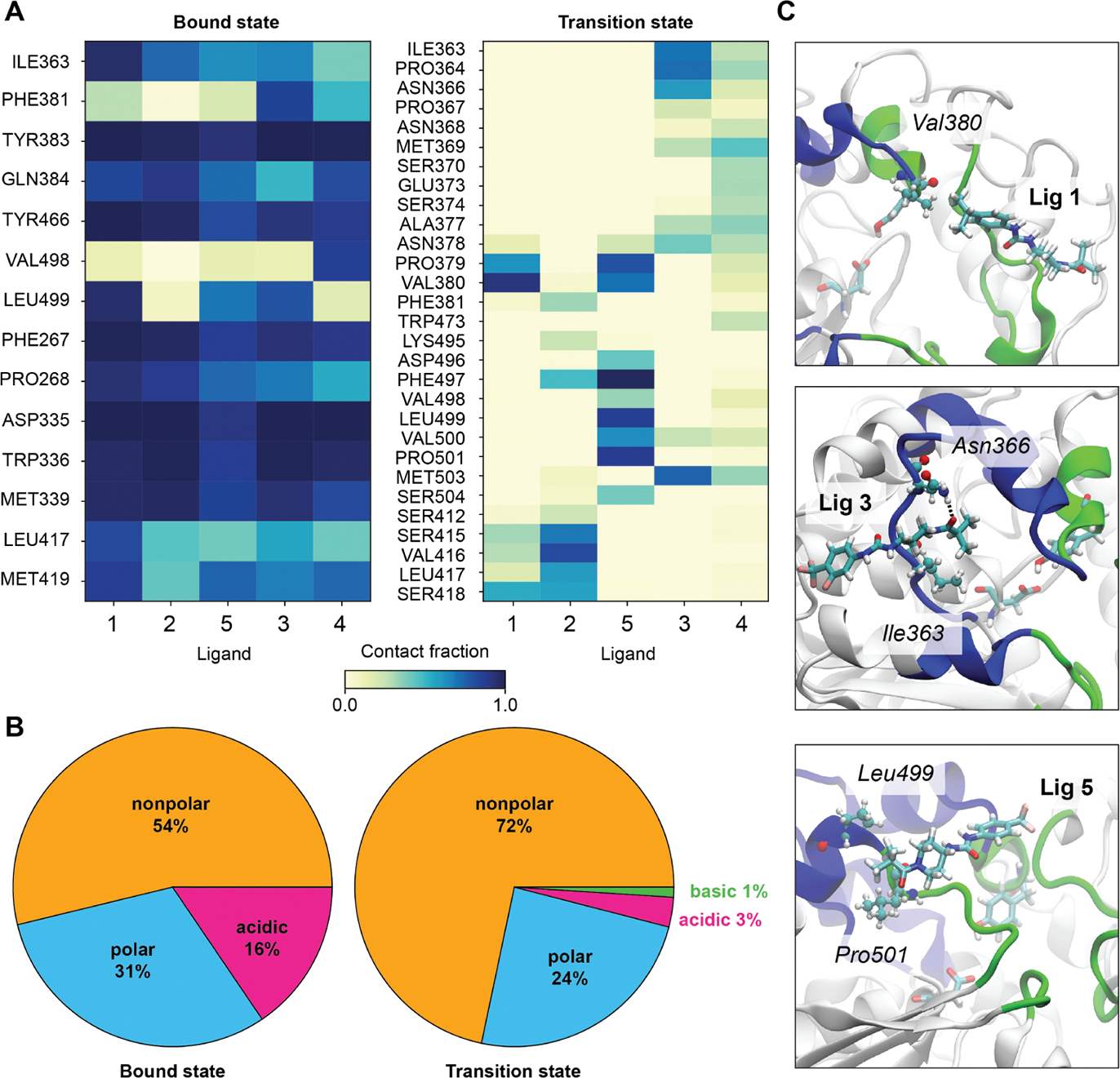
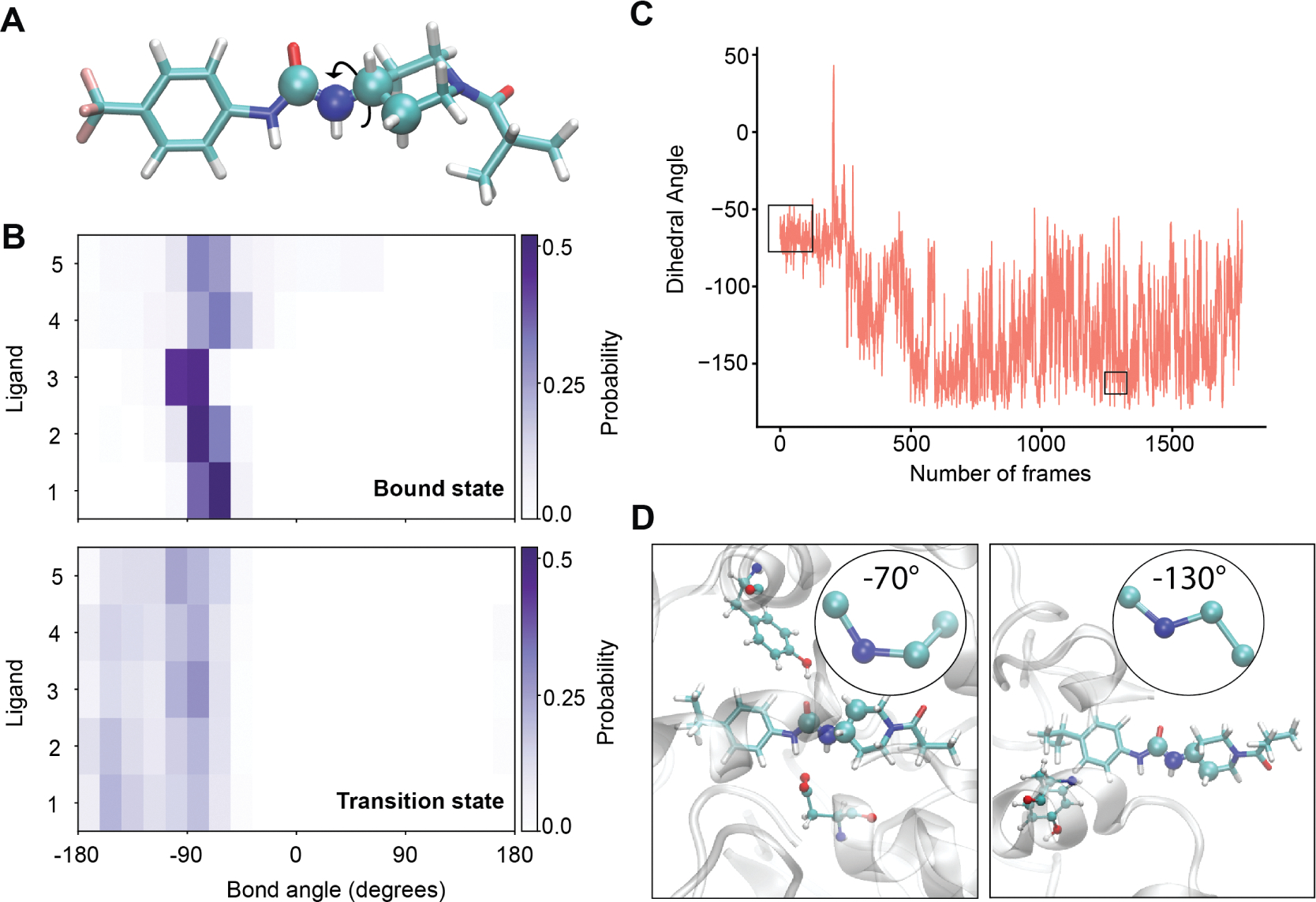
For many drug targets, it has been shown that the kinetics of drug binding (e.g., on rate and off rate) is more predictive of drug efficacy than thermodynamic quantities alone. This motivates the development of predictive computational models that can be used to optimize compounds on the basis of their kinetics. The structural details underpinning these computational models are found not only in the bound state but also in the short-lived ligand binding transition states. Although transition states cannot be directly observed experimentally due to their extremely short lifetimes, recent successes have demonstrated that modeling the ligand binding transition state is possible with the help of enhanced sampling molecular dynamics methods. Previously, we generated unbinding paths for an inhibitor of soluble epoxide hydrolase (sEH) with a residence time of 11 min. Here, we computationally modeled unbinding events with the weighted ensemble method REVO (resampling of ensembles by variation optimization) for five additional inhibitors of sEH with residence times ranging from 14.25 to 31.75 min, with average prediction accuracy within an order of magnitude. The unbinding ensembles are analyzed in detail, focusing on features of the ligand binding transition state ensembles (TSEs). We find that ligands with similar bound poses can show significant differences in their ligand binding TSEs, in terms of their spatial distribution and protein-ligand interactions. However, we also find similarities across the TSEs when examining more general features such as ligand degrees of freedom. Together these findings show significant challenges for rational, kinetics-based drug design.
Conflict of interest statement
Notes
The authors declare no competing financial interest.
Figures
References
-
- Greer J; Erickson JW; Baldwin JJ; Varney MD Application of the Three-Dimensional Structures of Protein Target Molecules in Structure-Based Drug Design. J. Med. Chem. 1994, 37, 1035–1054. - PubMed
-
- Rutenber EE; Stroud RM Binding of the anticancer drug ZD1694 to E. coli thymidylate synthase: Assessing specificity and affinity. Structure 1996, 4, 1317–1324. - PubMed
-
- Wlodawer A; Vondrasek J Inhibitors of HIV-1 protease: A major success of structure-assisted drug design. Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 249–284. - PubMed
-
- Clark DE What has computer-aided molecular design ever done for drug discovery? Expert Opin. Drug Discovery 2006, 1, 103–110. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
