

Targeted gene expression profiling predicts meningioma outcomes and radiotherapy responses
- PMID: 37944590
- PMCID: PMC11073469
- DOI: 10.1038/s41591-023-02586-z
Targeted gene expression profiling predicts meningioma outcomes and radiotherapy responses
Abstract
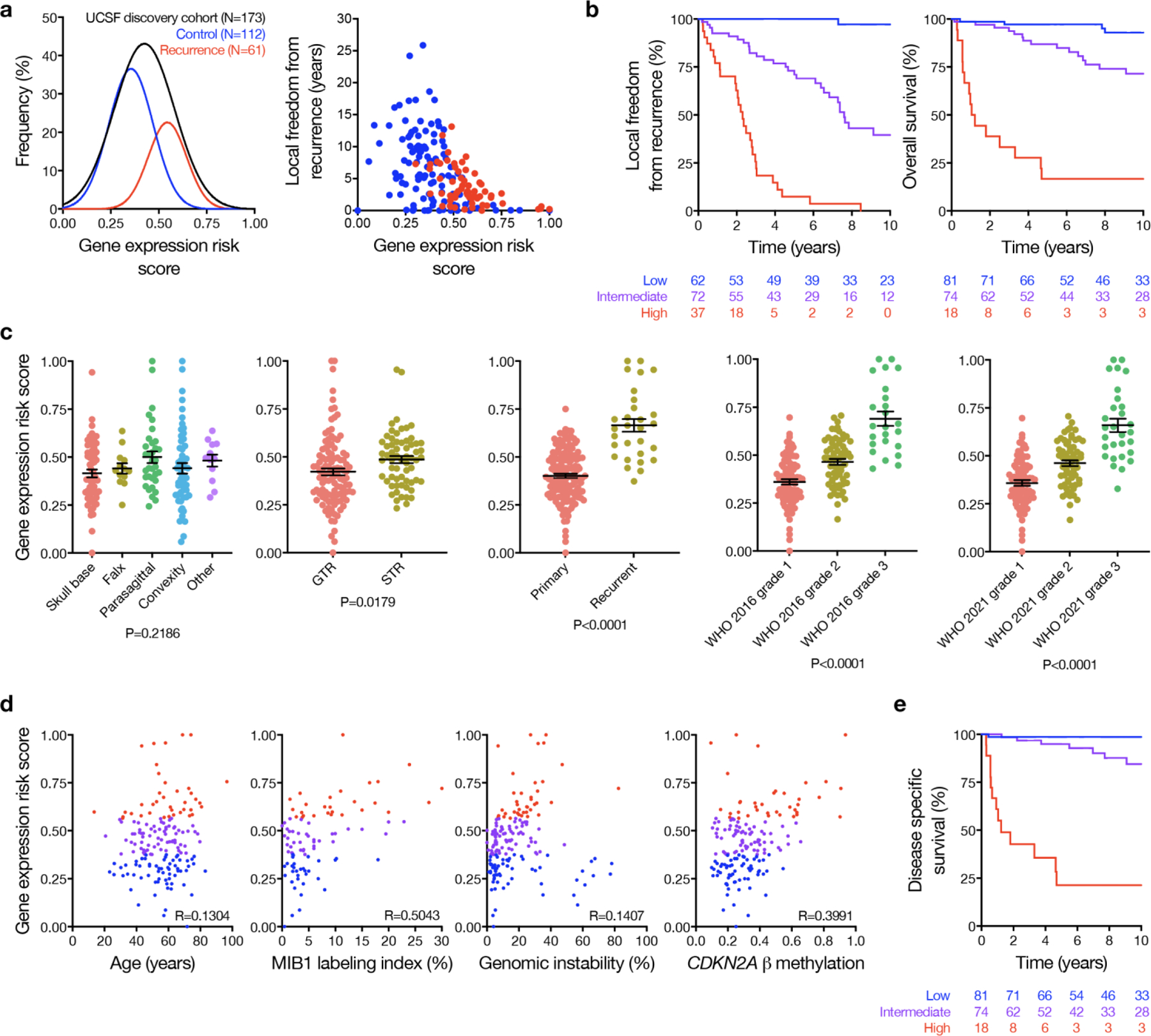
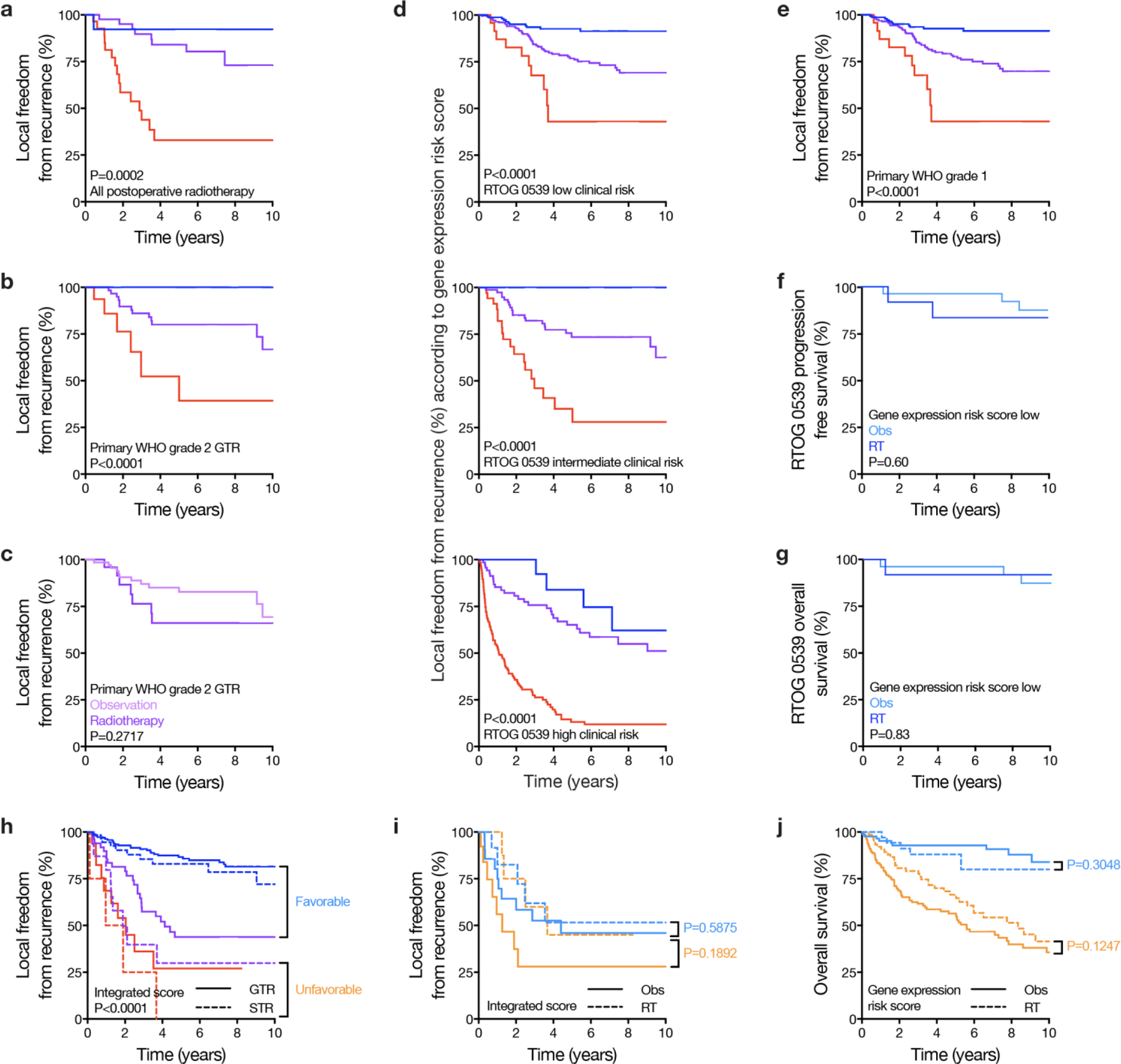
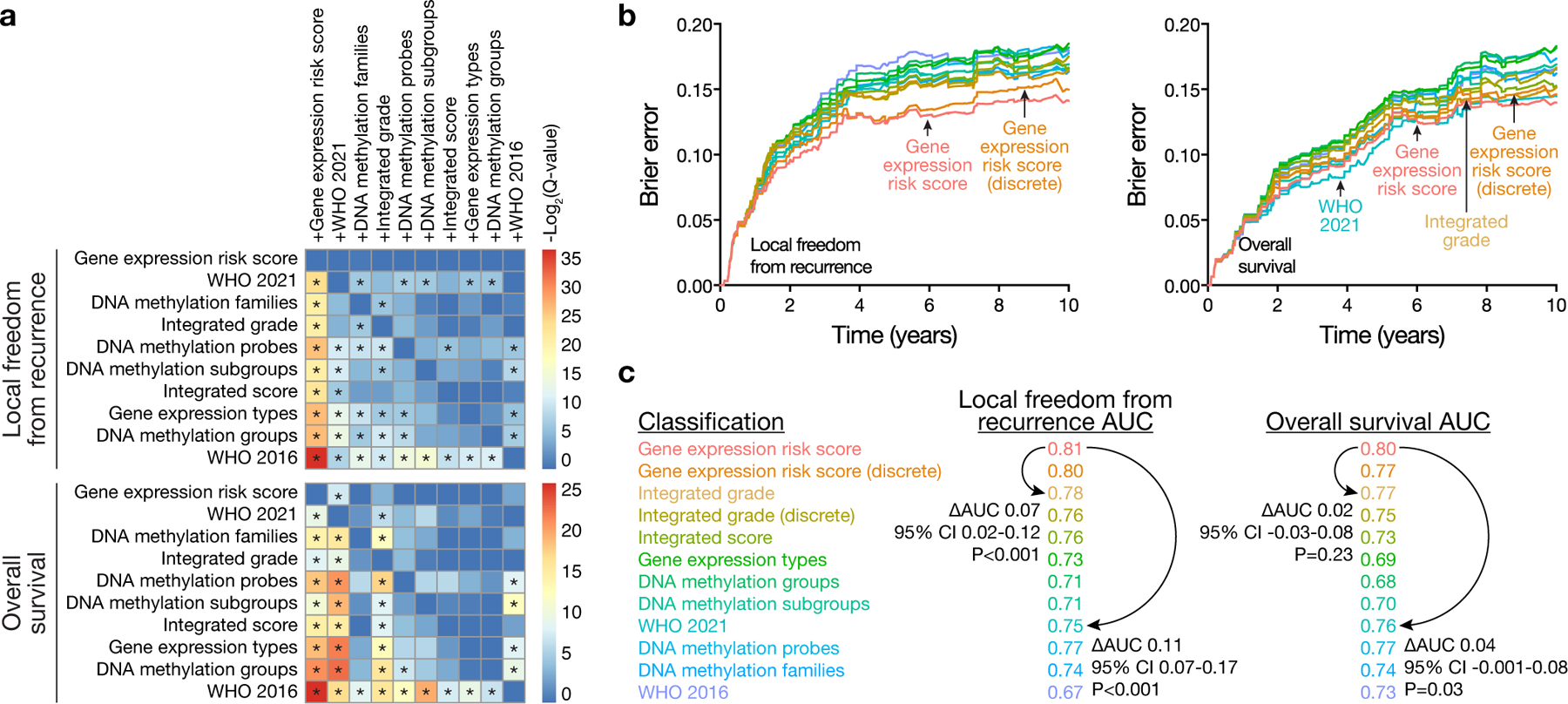
Surgery is the mainstay of treatment for meningioma, the most common primary intracranial tumor, but improvements in meningioma risk stratification are needed and indications for postoperative radiotherapy are controversial. Here we develop a targeted gene expression biomarker that predicts meningioma outcomes and radiotherapy responses. Using a discovery cohort of 173 meningiomas, we developed a 34-gene expression risk score and performed clinical and analytical validation of this biomarker on independent meningiomas from 12 institutions across 3 continents (N = 1,856), including 103 meningiomas from a prospective clinical trial. The gene expression biomarker improved discrimination of outcomes compared with all other systems tested (N = 9) in the clinical validation cohort for local recurrence (5-year area under the curve (AUC) 0.81) and overall survival (5-year AUC 0.80). The increase in AUC compared with the standard of care, World Health Organization 2021 grade, was 0.11 for local recurrence (95% confidence interval 0.07 to 0.17, P < 0.001). The gene expression biomarker identified meningiomas benefiting from postoperative radiotherapy (hazard ratio 0.54, 95% confidence interval 0.37 to 0.78, P = 0.0001) and suggested postoperative management could be refined for 29.8% of patients. In sum, our results identify a targeted gene expression biomarker that improves discrimination of meningioma outcomes, including prediction of postoperative radiotherapy responses.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests statement
MP has received honoraria for lectures, consultation, or advisory board participation from the following for-profit companies: Bayer, Bristol-Myers Squibb, Novartis, Gerson Lehrman Group (GLG), CMC Contrast, GlaxoSmithKline, Mundipharma, Roche, BMJ Journals, MedMedia, Astra Zeneca, AbbVie, Lilly, Medahead, Daiichi Sankyo, Sanofi, Merck Sharp & Dome, Tocagen, Adastra, Gan & Lee Pharmaceuticals. WCC and DRR are the inventors on patent PCT/US 21/70288 describing the use of targeted gene expression profiling to predict meningioma outcomes and radiotherapy responses. The remaining authors declare no competing interests.
Figures











Update of
-
Targeted gene expression profiling predicts meningioma outcomes and radiotherapy responses.Res Sq [Preprint]. 2023 Mar 20:rs.3.rs-2663611. doi: 10.21203/rs.3.rs-2663611/v1. Res Sq. 2023. Update in: Nat Med. 2023 Dec;29(12):3067-3076. doi: 10.1038/s41591-023-02586-z. PMID: 36993741 Free PMC article. Updated. Preprint.
