

Structural and non-coding variants increase the diagnostic yield of clinical whole genome sequencing for rare diseases
- PMID: 37946251
- PMCID: PMC10636885
- DOI: 10.1186/s13073-023-01240-0
Structural and non-coding variants increase the diagnostic yield of clinical whole genome sequencing for rare diseases
Abstract
Background: Whole genome sequencing is increasingly being used for the diagnosis of patients with rare diseases. However, the diagnostic yields of many studies, particularly those conducted in a healthcare setting, are often disappointingly low, at 25-30%. This is in part because although entire genomes are sequenced, analysis is often confined to in silico gene panels or coding regions of the genome.
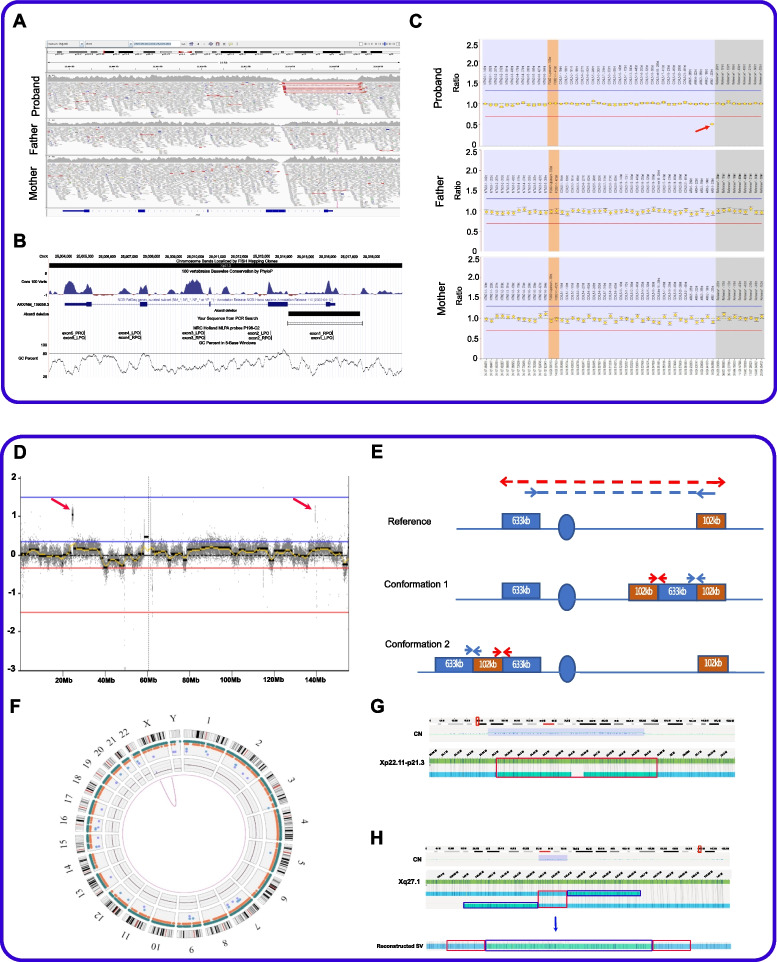
Methods: We undertook WGS on a cohort of 122 unrelated rare disease patients and their relatives (300 genomes) who had been pre-screened by gene panels or arrays. Patients were recruited from a broad spectrum of clinical specialties. We applied a bioinformatics pipeline that would allow comprehensive analysis of all variant types. We combined established bioinformatics tools for phenotypic and genomic analysis with our novel algorithms (SVRare, ALTSPLICE and GREEN-DB) to detect and annotate structural, splice site and non-coding variants.
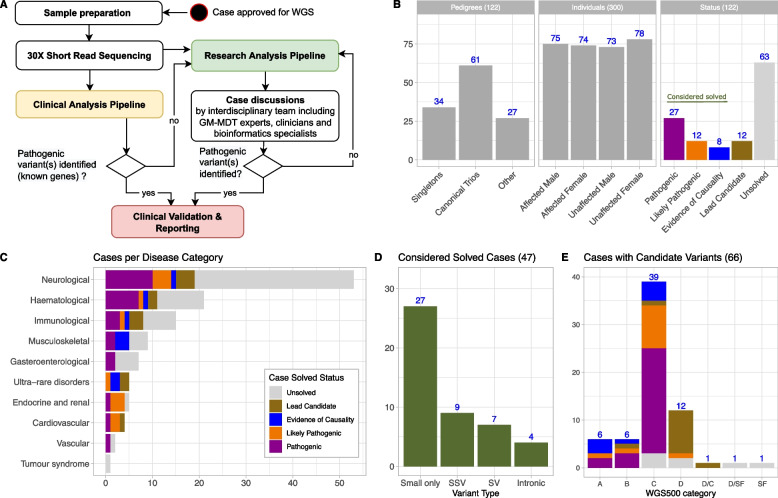
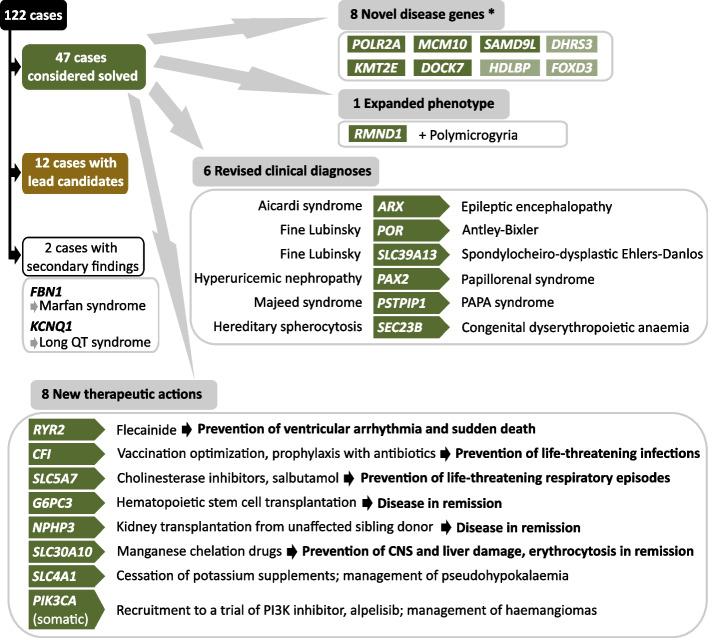
Results: Our diagnostic yield was 43/122 cases (35%), although 47/122 cases (39%) were considered solved when considering novel candidate genes with supporting functional data into account. Structural, splice site and deep intronic variants contributed to 20/47 (43%) of our solved cases. Five genes that are novel, or were novel at the time of discovery, were identified, whilst a further three genes are putative novel disease genes with evidence of causality. We identified variants of uncertain significance in a further fourteen candidate genes. The phenotypic spectrum associated with RMND1 was expanded to include polymicrogyria. Two patients with secondary findings in FBN1 and KCNQ1 were confirmed to have previously unidentified Marfan and long QT syndromes, respectively, and were referred for further clinical interventions. Clinical diagnoses were changed in six patients and treatment adjustments made for eight individuals, which for five patients was considered life-saving.
Conclusions: Genome sequencing is increasingly being considered as a first-line genetic test in routine clinical settings and can make a substantial contribution to rapidly identifying a causal aetiology for many patients, shortening their diagnostic odyssey. We have demonstrated that structural, splice site and intronic variants make a significant contribution to diagnostic yield and that comprehensive analysis of the entire genome is essential to maximise the value of clinical genome sequencing.
Keywords: Bioinformatics pipeline development; Clinical impact; Diagnostic yield; Genome sequencing; Non-coding; Pipeline optimisation; Rare diseases; Splice site variant; Structural variant.
© 2023. Crown.
Conflict of interest statement
JRH is a founder, director, paid consultant and shareholder of Nucleome Therapeutics.
SL reports consulting fees from Sanofi, GLG consulting, Atheneum and Rejuversen. He has received payment or honoraria for lectures, presentations, or educational events from Eisai, Prosigna, Roche, Pfizer, Novartis, Shionogi and Sanofi and was previously employed by Pfizer. He has received travel, accommodation or expenses from Pfizer, Roche, Synthon and Piqur Therapeutics and research funding from CRUK, Against Breast Cancer, Pathios Therapeutics and is cofounder of Mitox Therapeutics. His institution has received funding for clinical trials for which he is chief investigator or principle investigator from CRUK, Boehringer Ingelheim, Piqur Therapeutics, Astra Zeneca, Carrick Therapeutics, Sanofi, Merck KGaA, Synthon, Roche and Prostate Cancer UK.
GL is a founder and shareholder of Genomics PLC.
JP acknowledges the following: support for scientific meetings and honorariums for advisory work from Merck Serono, Novartis, Chugai, Alexion, Roche, Medimmune, Argenx, UCB, Mitsubishi, Amplo, Janssen, Sanofi; grants from Alexion, Roche, Medimmune, UCB, Amplo biotechnology, Argenx; patent ref P37347WO and licence agreement Numares multimarker MS diagnostics; shares in AstraZeneca; partial funding by highly specialised services NHS England. For CMS itself, support for advisory work and research grants from Argenx and Amplo biotechnology. None are a conflict for this work.
HHU has received research support or consultancy fees from Janssen, UCB Pharma, Eli Lilly, Bristol Myers Squibb BMS, OMass, Mestag, Mirobio, AbbVie and GSK.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- MR/M006824/1/MRC_/Medical Research Council/United Kingdom
- CH/1992001/6764/BHF_/British Heart Foundation/United Kingdom
- MC_UU_00029/3/MRC_/Medical Research Council/United Kingdom
- MC_UU_00016/1/MRC_/Medical Research Council/United Kingdom
- MR/T031670/1/MRC_/Medical Research Council/United Kingdom
- MC_UU_00008/6/MRC_/Medical Research Council/United Kingdom
- 102731/WT_/Wellcome Trust/United Kingdom
- 102731/Z/13/Z/WT_/Wellcome Trust/United Kingdom
- MC_PC_21044/MRC_/Medical Research Council/United Kingdom
- MC_UU_00016/14 /MRC_/Medical Research Council/United Kingdom
- MR/WO1761X/1/MRC_/Medical Research Council/United Kingdom
- MC_UU_00008/MRC_/Medical Research Council/United Kingdom
- MR/T014067/1/MRC_/Medical Research Council/United Kingdom
- MC_UU_00029/ 01-09/MRC_/Medical Research Council/United Kingdom
- 223,149/Z/21/Z/WT_/Wellcome Trust/United Kingdom
- MR/S007180/1/MRC_/Medical Research Council/United Kingdom
- 222096/Z/20/Z/WT_/Wellcome Trust/United Kingdom
- 203141/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- DH_/Department of Health/United Kingdom
- G0902418/MRC_/Medical Research Council/United Kingdom
- R6-388 / WT 100127/WT_/Wellcome Trust/United Kingdom
- MR/X001210/1/MRC_/Medical Research Council/United Kingdom
- 106,130/Z/14/Z/WT_/Wellcome Trust/United Kingdom
- MR/W01761X/1/MRC_/Medical Research Council/United Kingdom
- MR/R007748/1/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
