

A 15-year consolidated overview of data in over 6000 patients from the Transthyretin Amyloidosis Outcomes Survey (THAOS)
- PMID: 37946256
- PMCID: PMC10636983
- DOI: 10.1186/s13023-023-02962-5
A 15-year consolidated overview of data in over 6000 patients from the Transthyretin Amyloidosis Outcomes Survey (THAOS)
Abstract
Background: Transthyretin amyloidosis (ATTR amyloidosis) is a progressive, multisystemic, life-threatening disease resulting from the deposition of variant or wild-type (ATTRwt amyloidosis) transthyretin amyloid fibrils in various tissues and organs.
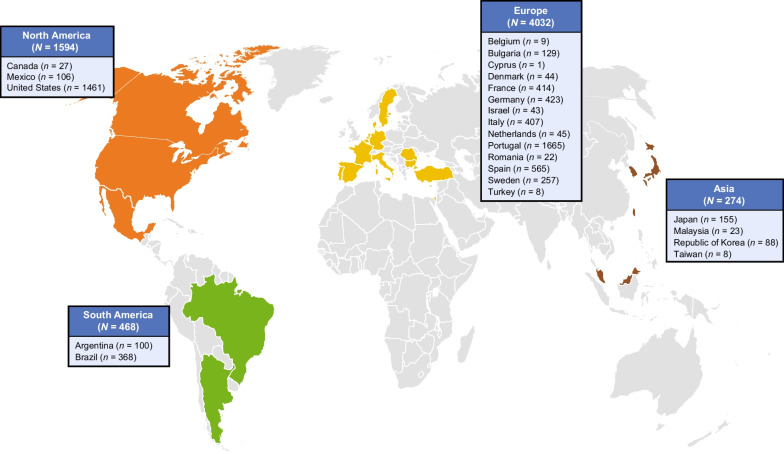
Methods: Established in 2007, the Transthyretin Amyloidosis Outcomes Survey (THAOS) is the largest ongoing, global, longitudinal, observational study of patients with ATTR amyloidosis, including both hereditary and wild-type disease, and asymptomatic carriers of pathogenic TTR mutations. This analysis describes the baseline characteristics of symptomatic patients and asymptomatic gene carriers enrolled in THAOS since its inception in 2007 (data cutoff: August 1, 2022), providing a consolidated overview of 15-year data from the THAOS registry.
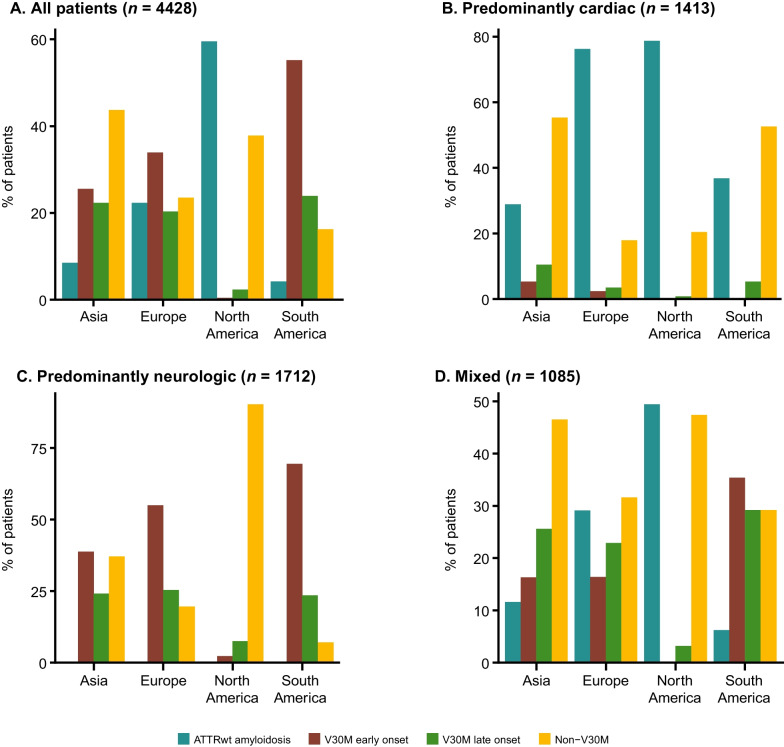
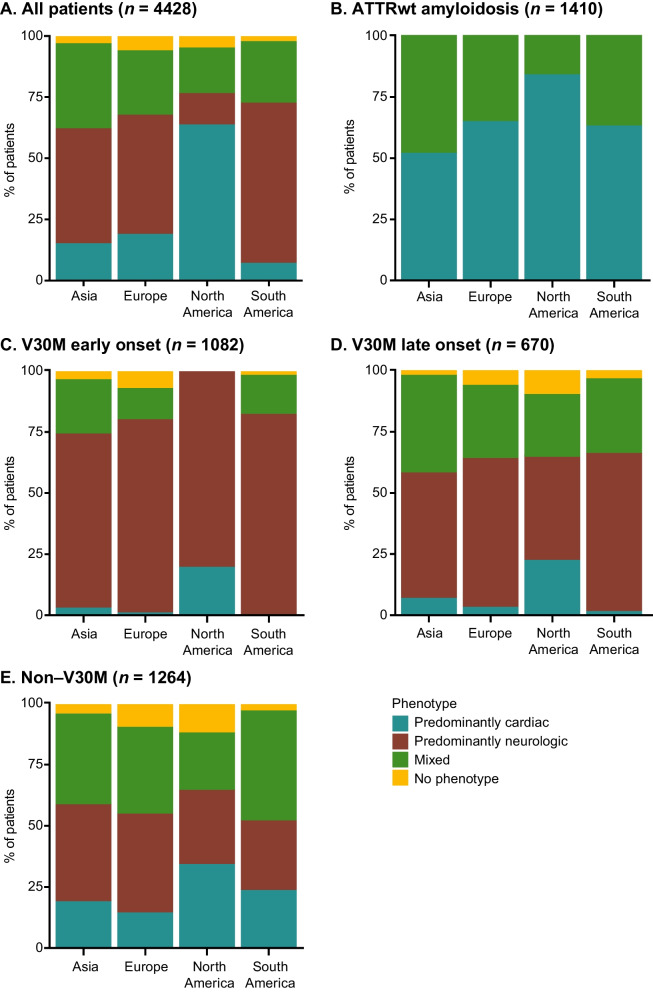
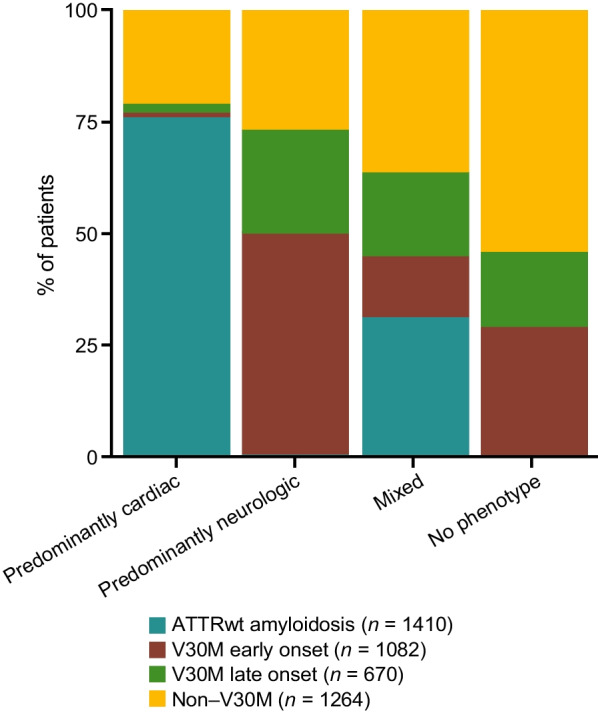
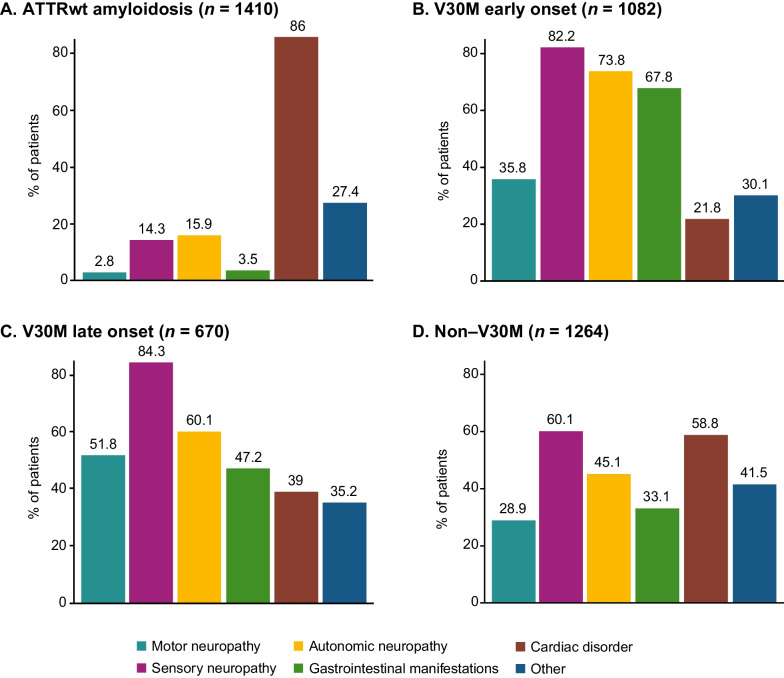
Results: This analysis included 4428 symptomatic patients and 1707 asymptomatic gene carriers. The majority of symptomatic patients were male (70.8%) with a mean (standard deviation [SD]) age at symptom onset of 56.6 (17.9) years. Compared with the 14-year analysis, V30M remained the most prevalent genotype in Europe (62.2%), South America (78.6%), and Japan (74.2%) and ATTRwt remained most common in North America (56.2%). Relative to the 14-year analysis, there was an increase of mixed phenotype (from 16.6 to 24.5%) and a reduction of predominantly cardiac phenotype (from 40.7 to 31.9%). The proportion of patients with predominantly neurologic phenotype remained stable (from 40.1 to 38.7%). Asymptomatic gene carriers were 58.5% female with a mean age at enrollment of 41.9 years (SD 15.5).
Conclusions: This overview of > 6000 patients enrolled over 15 years in THAOS represents the largest registry analysis of ATTR amyloidosis to date and continues to emphasize the genotypic and phenotypic heterogeneity of the disease. Nearly a quarter of the symptomatic population within THAOS was mixed phenotype, underscoring the need for multidisciplinary management of ATTR amyloidosis.
Trial registration: ClinicalTrials.gov Identifier: NCT00628745.
Keywords: Amyloidosis; Cardiomyopathy; Polyneuropathy; Registry; Transthyretin.
© 2023. The Author(s).
Conflict of interest statement
Luca Gentile reports travel grants from Kedrion and CSL Behring to attend scientific meetings. Teresa Coelho reports serving as a medical advisor for Pfizer and receiving funding for scientific meeting expenses (travel, accommodation and registration) from Pfizer. Angela Dispenzieri reports research grants from Celgene, Millennium, Pfizer, Janssen and Alnylam; funding from Pfizer for meeting expenses (travel); and attending advisory boards for Akcea and Intellia. Isabel Conceição reports consulting fees and funding for scientific meeting expenses (travel, accommodation and registration) from Pfizer. Márcia Waddington-Cruz reports research funding, consulting fees and travel support for advisory boards and meetings from FoldRx Pharmaceuticals and Pfizer. Arnt V. Kristen reports reimbursement for study visits from Pfizer during the conduct of the study. Jonas Wixner reports consulting fees and travel support for lectures and advisory boards from Pfizer and Alnylam; and consulting fees from Akcea and Intellia. Igor Diemberger received speaker fees from Biotronik, Boston Scientific, Daiichi Sankyo, Medtronic, Pfizer and Philips outside the submitted work. Juan Gonzalez-Moreno reports speaker fees from Pfizer, Alnylam and Akcea. Eve Cariou has no conflicts to report. Mathew S. Maurer reports grants from Pfizer during the conduct of the study; grants and personal fees from Pfizer, Eidos, Prothena and Ionis; grants from Alnylam; and personal fees from GSK and Akcea outside the submitted work. Violaine Planté-Bordeneuve reports serving as a medical advisor for Pfizer, Alnylam, Eidos and Ionis. Pablo Garcia-Pavia reports speaking fees from Pfizer, Eidos, Alnylam and Akcea; consulting fees from Pfizer, Eidos, Neurimmune, Alnylam, Prothena and Akcea; and research/educational support to his institution from Pfizer, Eidos and Alynylam. Ivailo Tournev reports speakers bureau/lecturer fees from Ewopharma, Genesis Pharma, Pfizer, Roche and Sanofi. Jose Gonzalez-Costello reports attending advisory boards for Pfizer and Alnylam. Alejandra González-Duarte reports financial support from Alnylam and Pfizer for her role as a principal investigator in studies sponsored by these entities, and as a consultant and speaker. Martha Grogan reports grants and advisory board and consultancy fees paid to her institution from Alnylam, Eidos, Prothena and Pfizer. Anna Mazzeo reports financial grants (honoraria and speaking) from Ackea, Alnylam, and Pfizer. Doug Chapman, Pritam Gupta, Oliver Glass, and Leslie Amass are current or former employees of Pfizer and hold stock and/or stock options in Pfizer.
Figures
References
Publication types
MeSH terms
Substances
Supplementary concepts
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
