

Inhibition of CARM1-Mediated Methylation of ACSL4 Promotes Ferroptosis in Colorectal Cancer
- PMID: 37946697
- PMCID: PMC10754121
- DOI: 10.1002/advs.202303484
Inhibition of CARM1-Mediated Methylation of ACSL4 Promotes Ferroptosis in Colorectal Cancer
Abstract
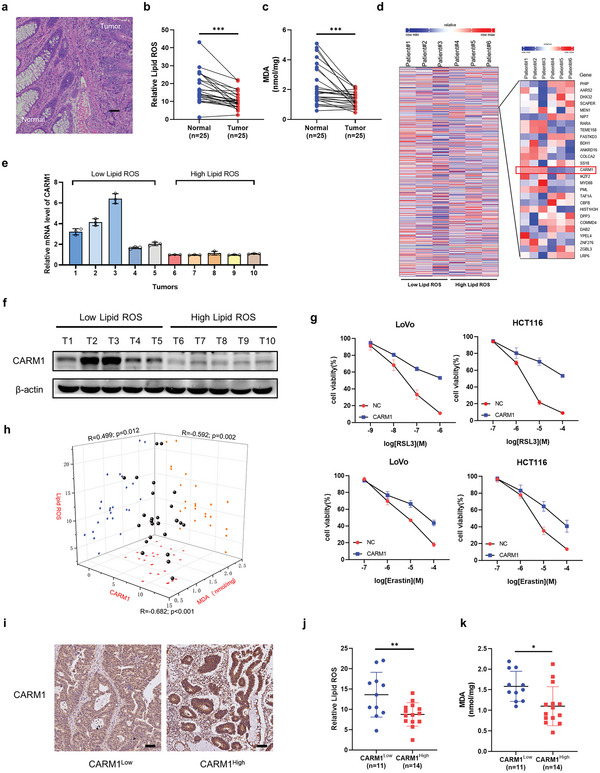
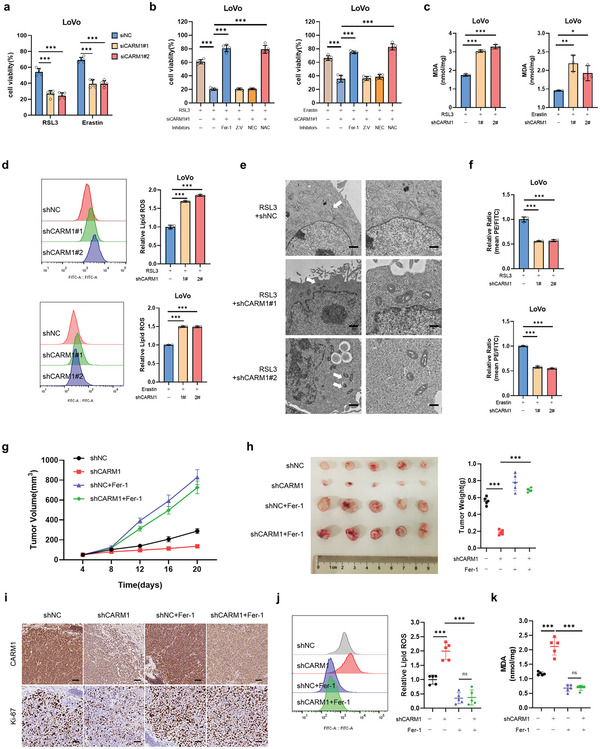
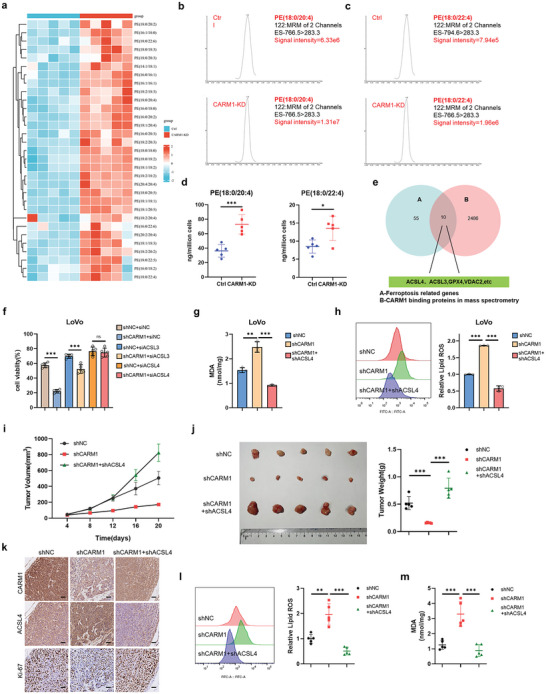
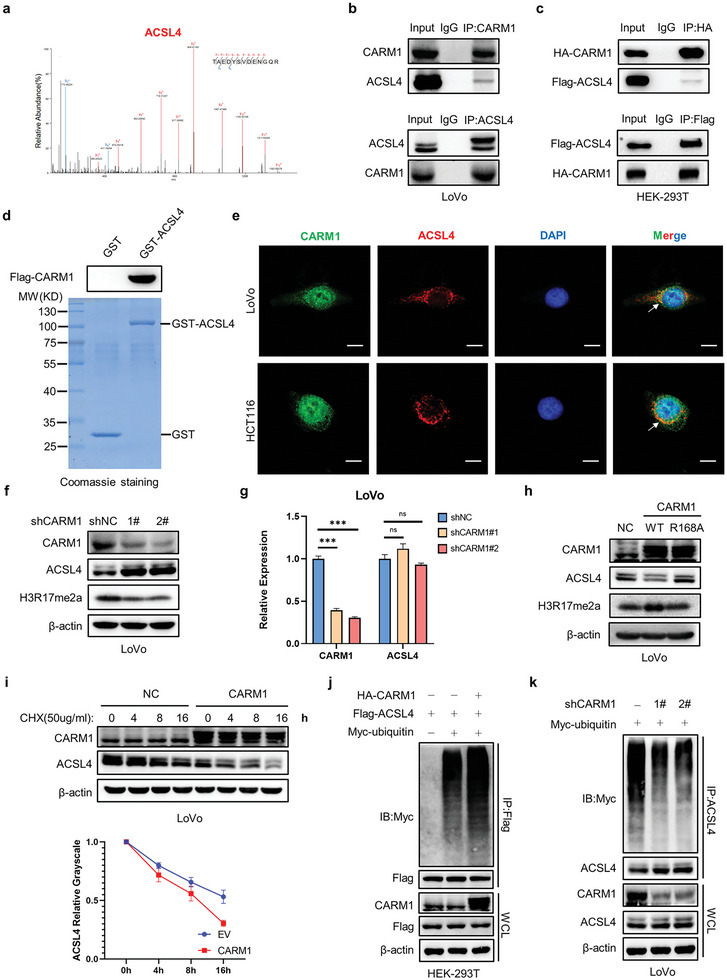
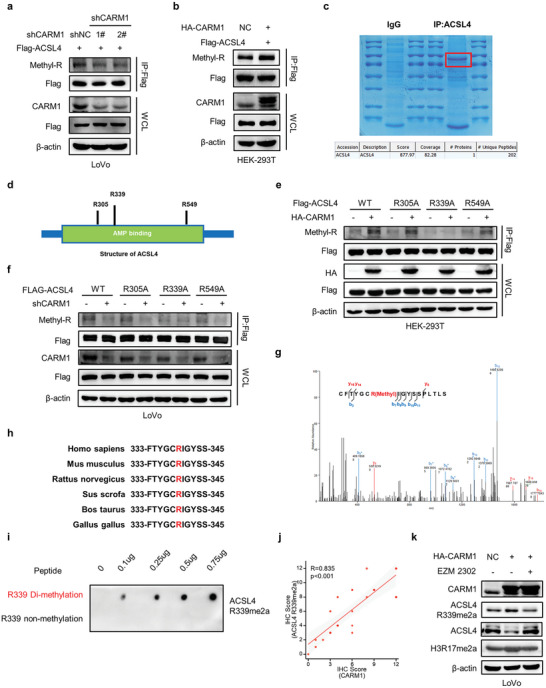
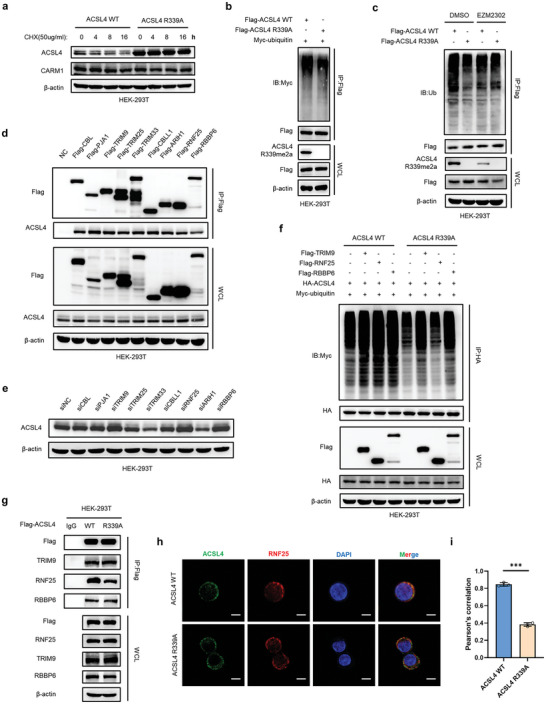
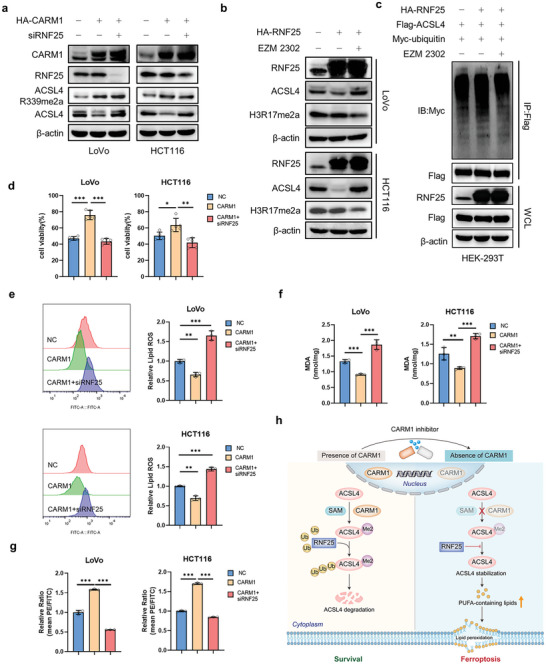
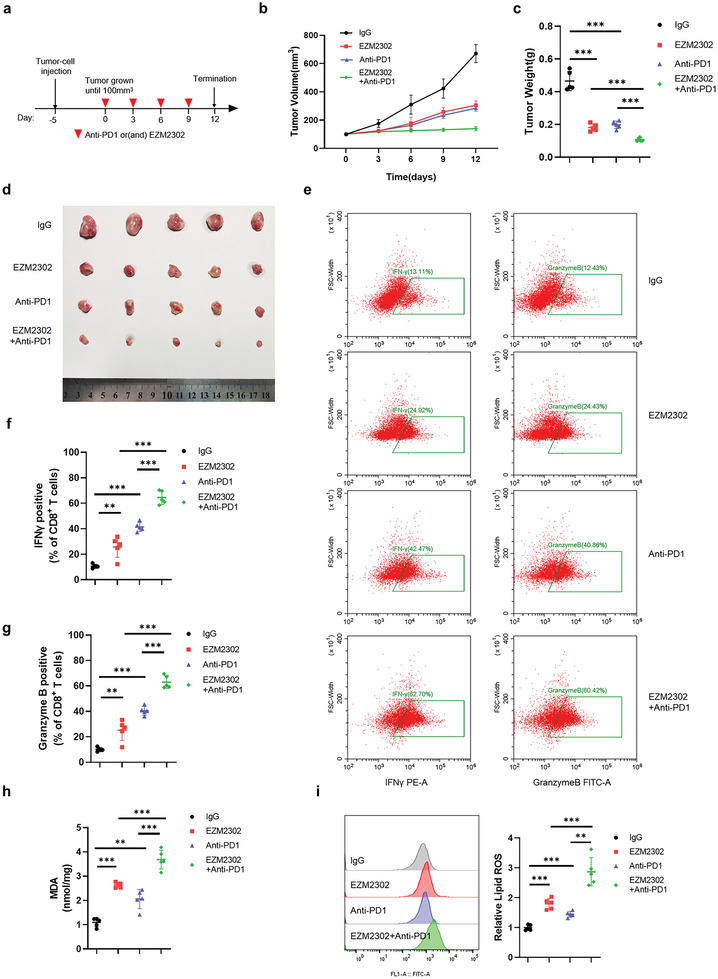
Ferroptosis, which is caused by iron-dependent accumulation of lipid peroxides, is an emerging form of regulated cell death and is considered a potential target for cancer therapy. However, the regulatory mechanisms underlying ferroptosis remain unclear. This study defines a distinctive role of ferroptosis. Inhibition of CARM1 can increase the sensitivity of tumor cells to ferroptosis inducers in vitro and in vivo. Mechanistically, it is found that ACSL4 is methylated by CARM1 at arginine 339 (R339). Furthermore, ACSL4 R339 methylation promotes RNF25 binding to ACSL4, which contributes to the ubiquitylation of ACSL4. The blockade of CARM1 facilitates ferroptosis and effectively enhances ferroptosis-associated cancer immunotherapy. Overall, this study demonstrates that CARM1 is a critical contributor to ferroptosis resistance and highlights CARM1 as a candidate therapeutic target for improving the effects of ferroptosis-based antitumor therapy.
Keywords: ACSL4 methylation; CARM1; ferroptosis.
© 2023 The Authors. Advanced Science published by Wiley-VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Friedmann Angeli J. P., Schneider M., Proneth B., Tyurina Y. Y., Tyurin V. A., Hammond V. J., Herbach N., Aichler M., Walch A., Eggenhofer E., Basavarajappa D., Rådmark O., Kobayashi S., Seibt T., Beck H., Neff F., Esposito I., Wanke R., Förster H., Yefremova O., Heinrichmeyer M., Bornkamm G. W., Geissler E. K., Thomas S. B., Stockwell B. R., O'donnell V. B., Kagan V. E., Schick J. A., Conrad M., Nat. Cell Biol. 2014, 16, 1180. - PMC - PubMed
-
- Ahola S., Rivera P. M., Hermans S., Chandragiri S., Giavalisco P., Nolte H., Langer T., Cell Metab. 2022, 34, 1875. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases