

Leveraging information between multiple population groups and traits improves fine-mapping resolution
- PMID: 37949886
- PMCID: PMC10638399
- DOI: 10.1038/s41467-023-43159-5
Leveraging information between multiple population groups and traits improves fine-mapping resolution
Abstract
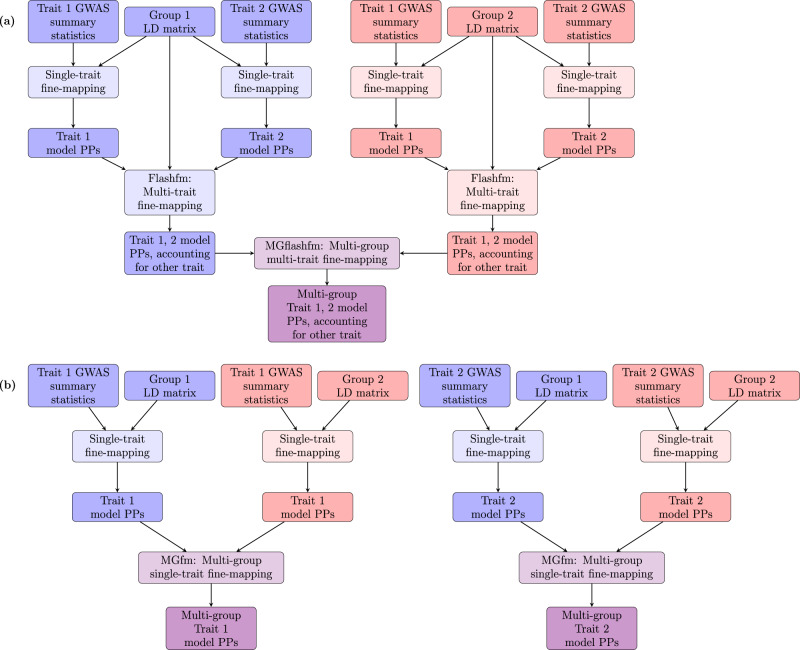
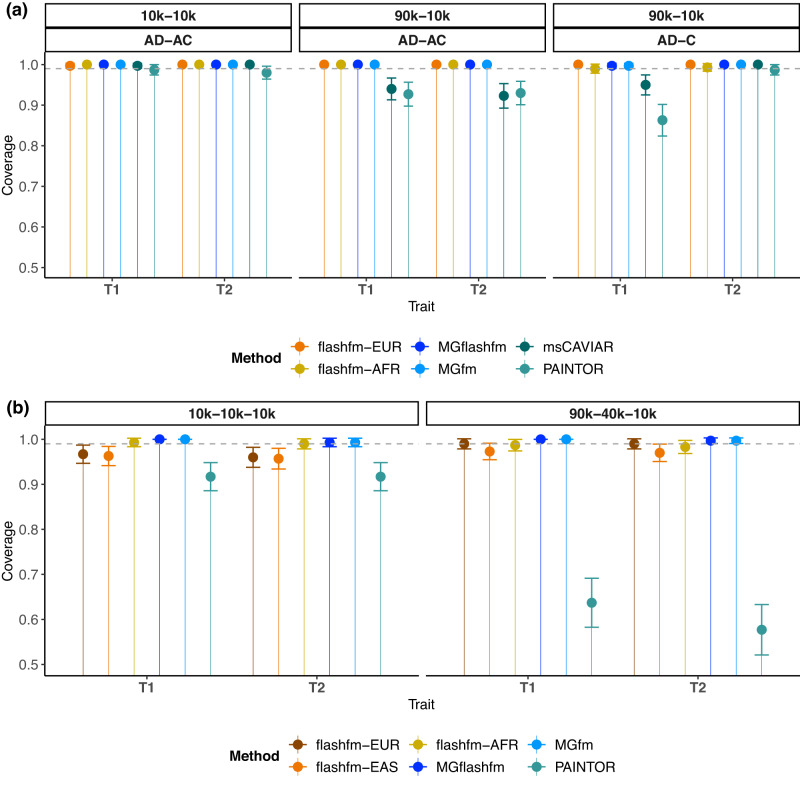
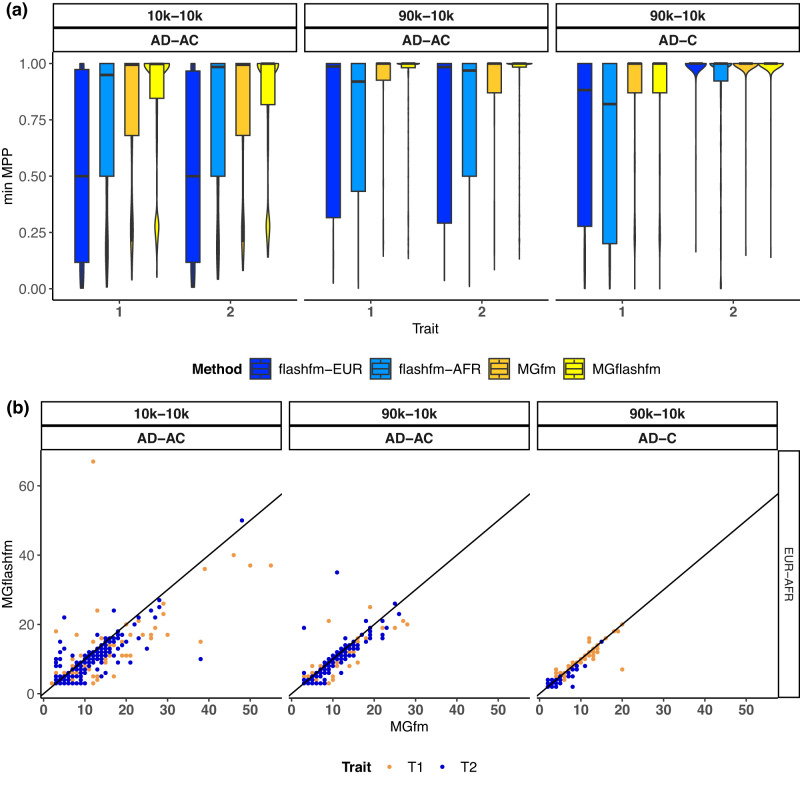
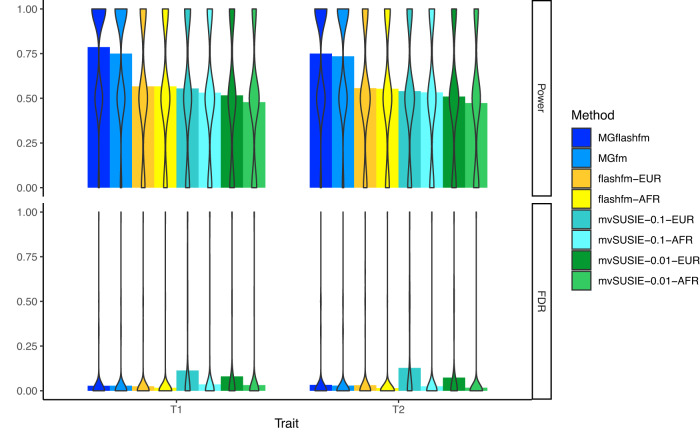
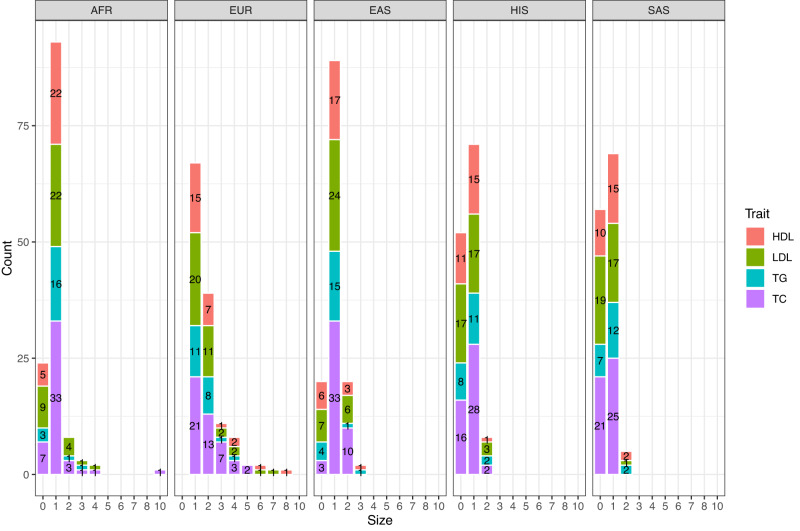
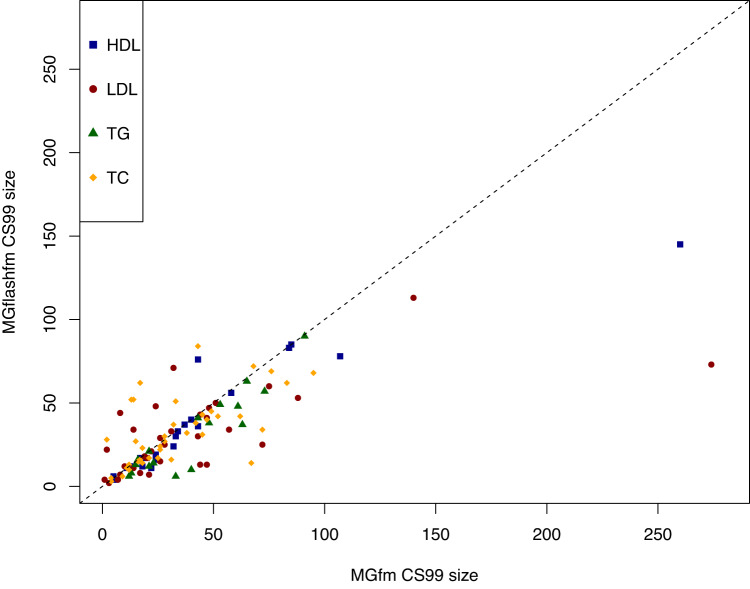
Statistical fine-mapping helps to pinpoint likely causal variants underlying genetic association signals. Its resolution can be improved by (i) leveraging information between traits; and (ii) exploiting differences in linkage disequilibrium structure between diverse population groups. Using association summary statistics, MGflashfm jointly fine-maps signals from multiple traits and population groups; MGfm uses an analogous framework to analyse each trait separately. We also provide a practical approach to fine-mapping with out-of-sample reference panels. In simulation studies we show that MGflashfm and MGfm are well-calibrated and that the mean proportion of causal variants with PP > 0.80 is above 0.75 (MGflashfm) and 0.70 (MGfm). In our analysis of four lipids traits across five population groups, MGflashfm gives a median 99% credible set reduction of 10.5% over MGfm. MGflashfm and MGfm only require summary level data, making them very useful fine-mapping tools in consortia efforts where individual-level data cannot be shared.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
