

Exploring the disruption of SARS-CoV-2 RBD binding to hACE2
- PMID: 37954960
- PMCID: PMC10635427
- DOI: 10.3389/fchem.2023.1276760
Exploring the disruption of SARS-CoV-2 RBD binding to hACE2
Abstract
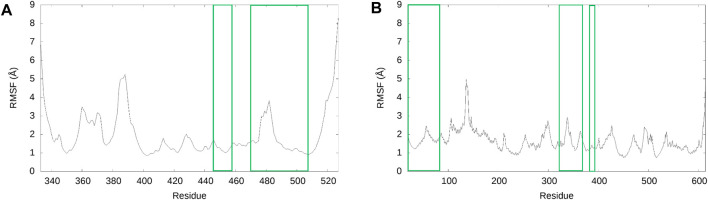
The COVID-19 pandemic was declared due to the spread of the novel coronavirus, SARS-CoV-2. Viral infection is caused by the interaction between the SARS-CoV-2 receptor binding domain (RBD) and the human ACE2 receptor (hACE2). Previous computational studies have identified repurposed small molecules that target the RBD, but very few have screened drugs in the RBD-hACE2 interface. When studies focus solely on the binding affinity between the drug and the RBD, they ignore the effect of hACE2, resulting in an incomplete analysis. We screened ACE inhibitors and previously identified SARS-CoV-2 inhibitors for binding to the RBD-hACE2 interface, and then conducted 500 ns of unrestrained molecular dynamics (MD) simulations of fosinopril, fosinoprilat, lisinopril, emodin, diquafosol, and physcion bound to the interface to assess the binding characteristics of these ligands. Based on MM-GBSA analysis, all six ligands bind favorably in the interface and inhibit the RBD-hACE2 interaction. However, when we repeat our simulation by first binding the drug to the RBD before interacting with hACE2, we find that fosinopril, fosinoprilat, and lisinopril result in a strongly interacting trimeric complex (RBD-drug-hACE2). Hydrogen bonding and pairwise decomposition analyses further suggest that fosinopril is the best RBD inhibitor. However, when lisinopril is bound, it stabilizes the trimeric complex and, therefore, is not an ideal potential drug candidate. Overall, these results reveal important atomistic interactions critical to the binding of the RBD to hACE2 and highlight the significance of including all protein partners in the evaluation of a potential drug candidate.
Keywords: ACE inhibitors; COVID-19; MM-GBSA; SARS-CoV-2; human ACE2 receptor; molecular dynamics; receptor binding domain; repurposed drugs.
Copyright © 2023 Carter, Airas, Gladden, Miller and Parish.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures






References
-
- Al-Karmalawy A. A., Dahab M. A., Metwaly A. M., Elhady S. S., Elkaeed E. B., Eissa I. H., et al. (2021). Molecular docking and dynamics simulation revealed the potential inhibitory activity of ACEIs against SARS-CoV-2 targeting the hACE2 receptor. Front. Chem. 9, 661230. 10.3389/fchem.2021.661230 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous