

Cyclin D-CDK4 Disulfide Bond Attenuates Pulmonary Vascular Cell Proliferation
- PMID: 37955182
- PMCID: PMC10699508
- DOI: 10.1161/CIRCRESAHA.122.321836
Cyclin D-CDK4 Disulfide Bond Attenuates Pulmonary Vascular Cell Proliferation
Abstract
Background: Pulmonary hypertension (PH) is a chronic vascular disease characterized, among other abnormalities, by hyperproliferative smooth muscle cells and a perturbed cellular redox and metabolic balance. Oxidants induce cell cycle arrest to halt proliferation; however, little is known about the redox-regulated effector proteins that mediate these processes. Here, we report a novel kinase-inhibitory disulfide bond in cyclin D-CDK4 (cyclin-dependent kinase 4) and investigate its role in cell proliferation and PH.
Methods: Oxidative modifications of cyclin D-CDK4 were detected in human pulmonary arterial smooth muscle cells and human pulmonary arterial endothelial cells. Site-directed mutagenesis, tandem mass-spectrometry, cell-based experiments, in vitro kinase activity assays, in silico structural modeling, and a novel redox-dead constitutive knock-in mouse were utilized to investigate the nature and definitively establish the importance of CDK4 cysteine modification in pulmonary vascular cell proliferation. Furthermore, the cyclin D-CDK4 oxidation was assessed in vivo in the pulmonary arteries and isolated human pulmonary arterial smooth muscle cells of patients with pulmonary arterial hypertension and in 3 preclinical models of PH.
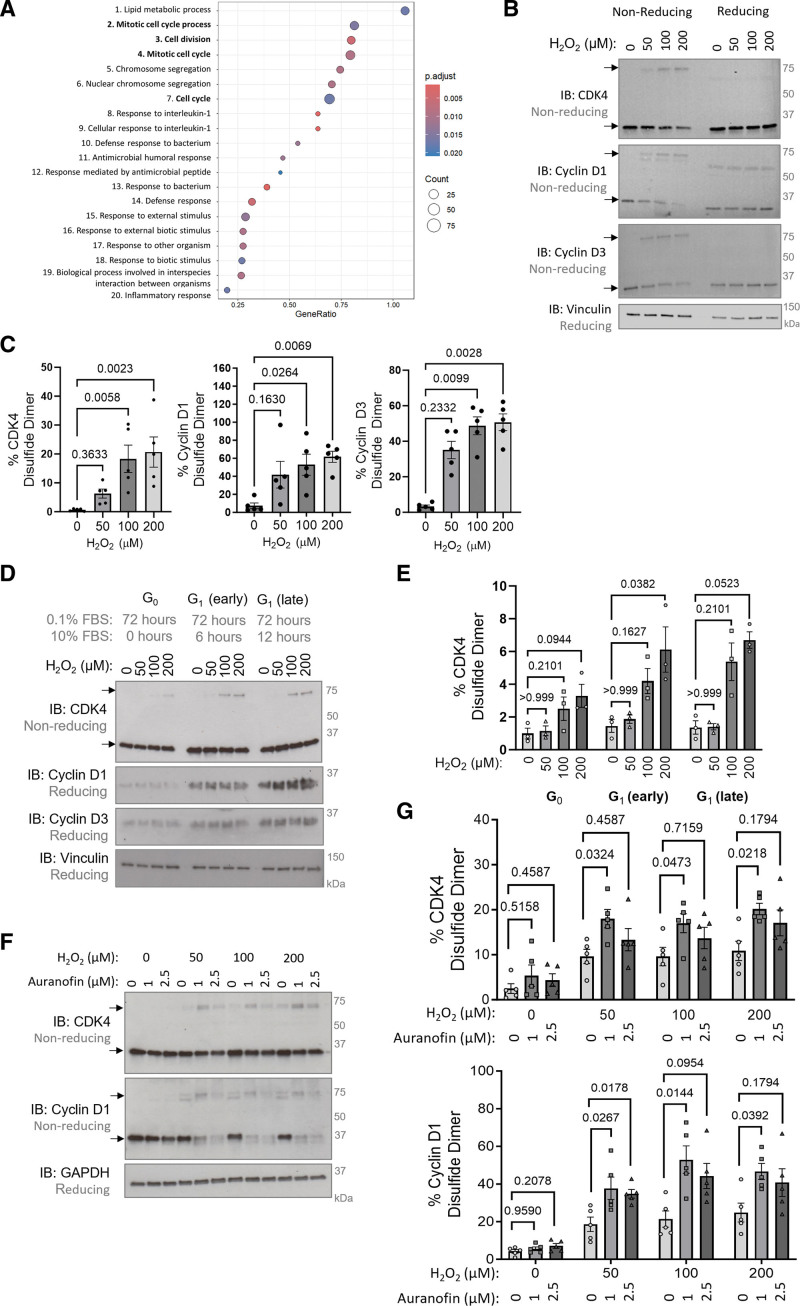
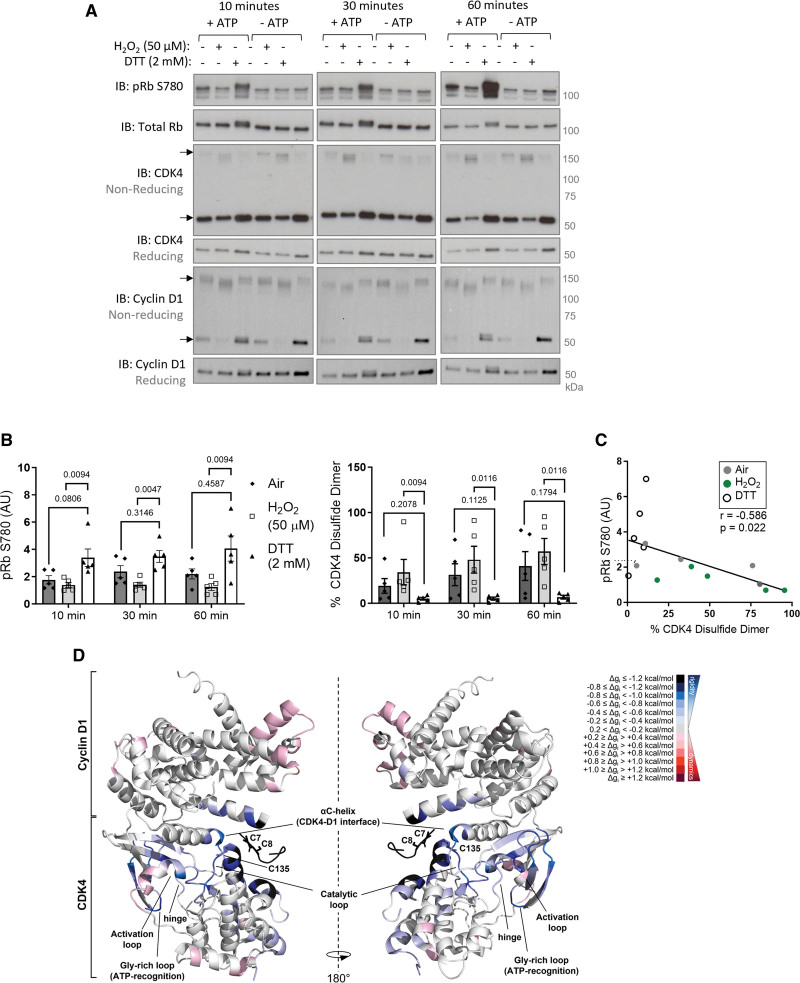
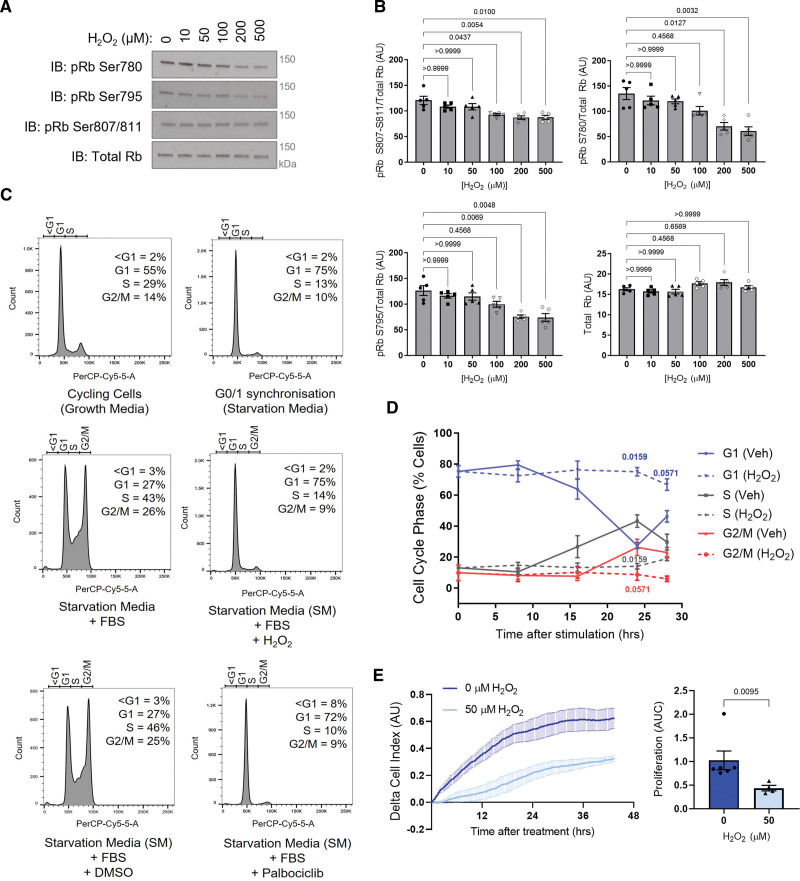
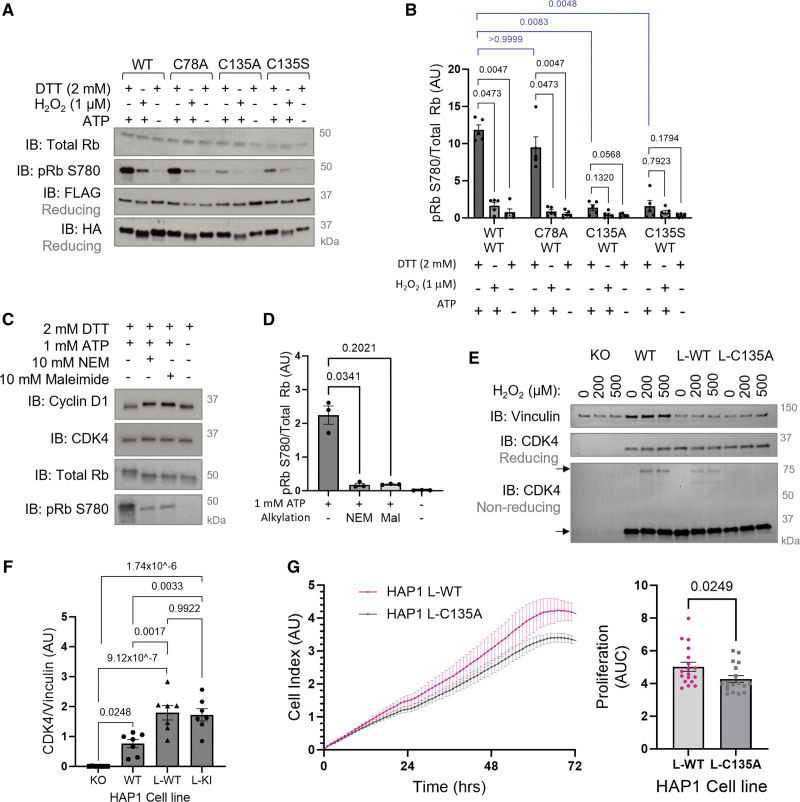
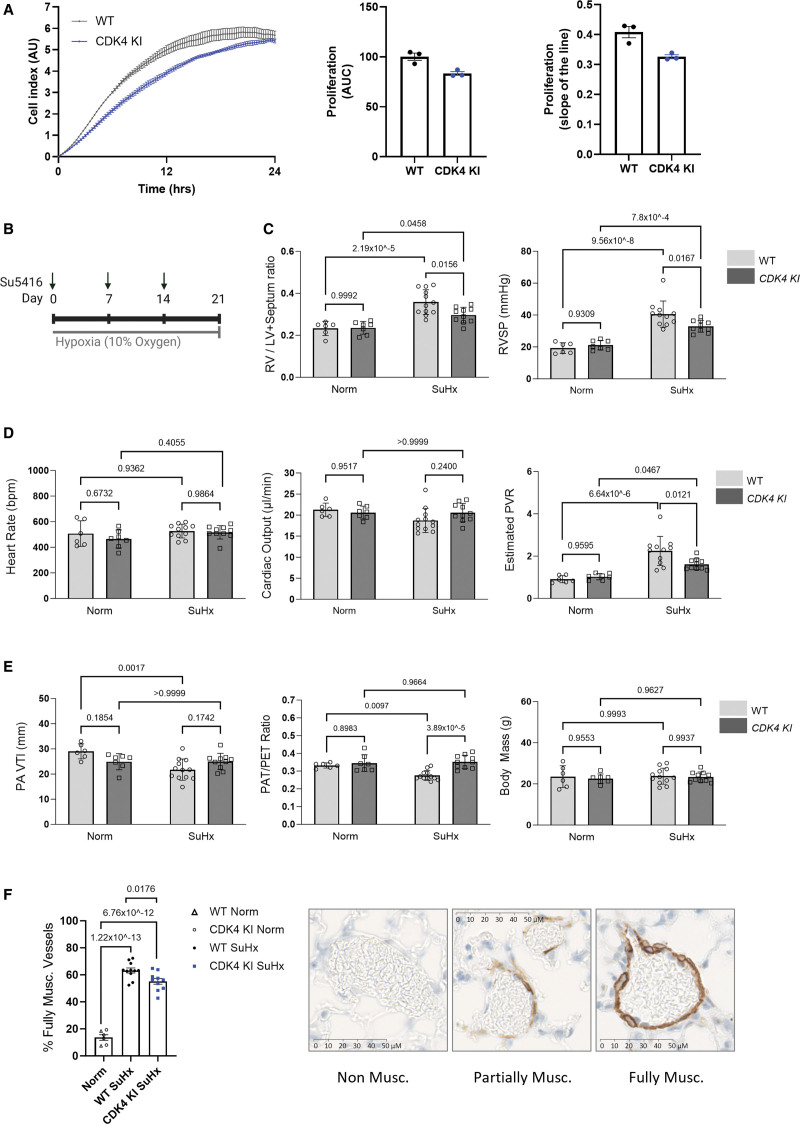
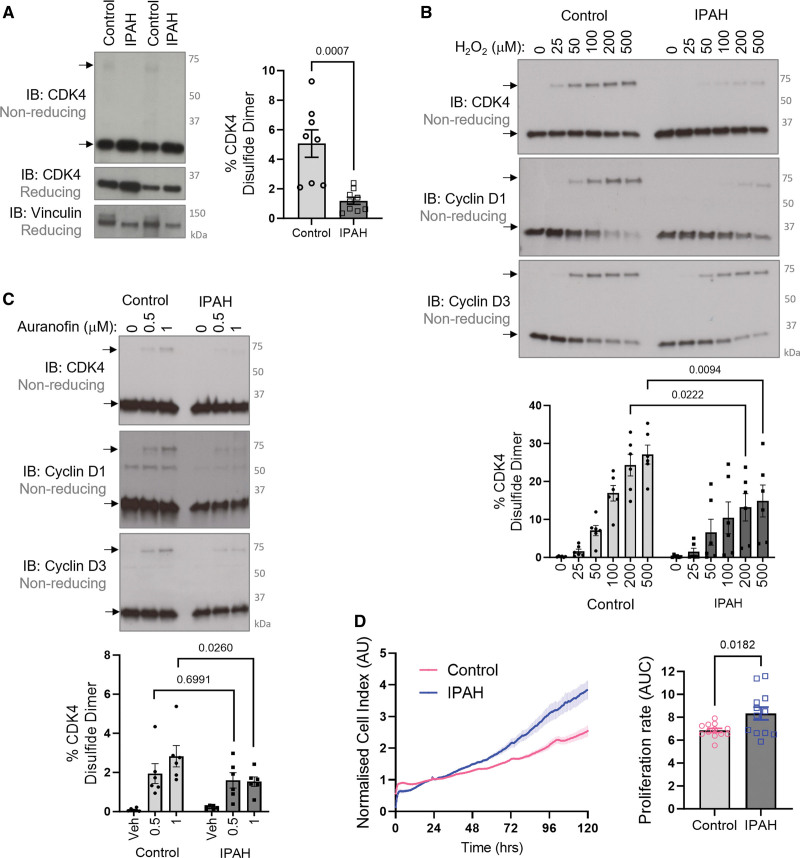
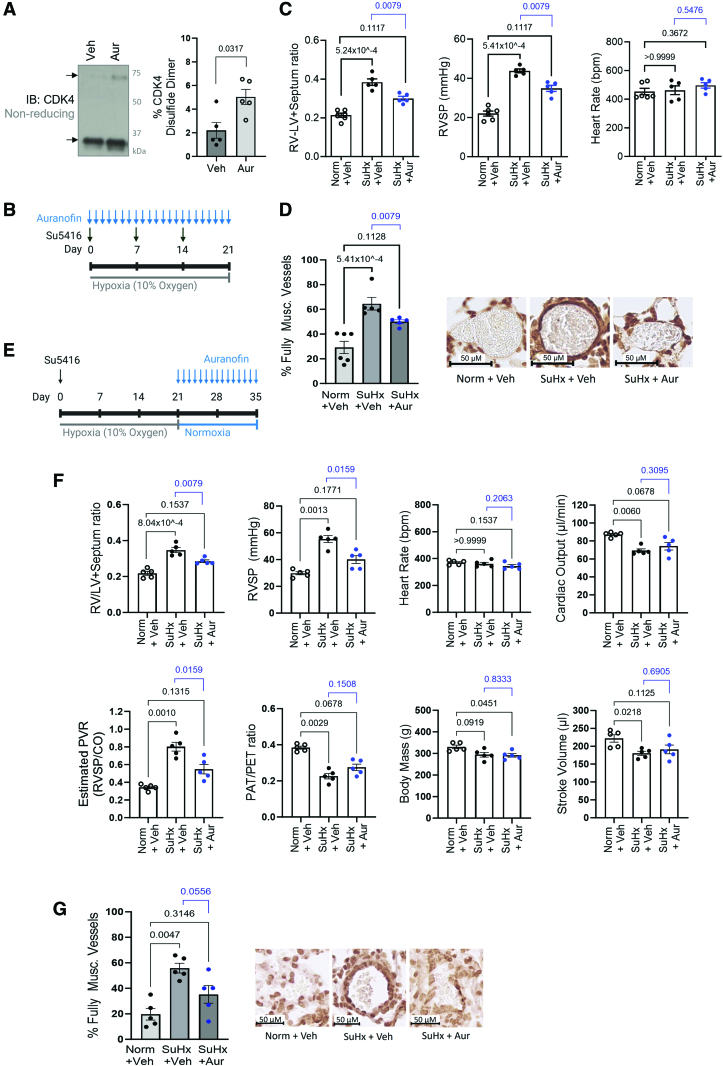
Results: Cyclin D-CDK4 forms a reversible oxidant-induced heterodimeric disulfide dimer between C7/8 and C135, respectively, in cells in vitro and in pulmonary arteries in vivo to inhibit cyclin D-CDK4 kinase activity, decrease Rb (retinoblastoma) protein phosphorylation, and induce cell cycle arrest. Mutation of CDK4 C135 causes a kinase-impaired phenotype, which decreases cell proliferation rate and alleviates disease phenotype in an experimental mouse PH model, suggesting this cysteine is indispensable for cyclin D-CDK4 kinase activity. Pulmonary arteries and human pulmonary arterial smooth muscle cells from patients with pulmonary arterial hypertension display a decreased level of CDK4 disulfide, consistent with CDK4 being hyperactive in human pulmonary arterial hypertension. Furthermore, auranofin treatment, which induces the cyclin D-CDK4 disulfide, attenuates disease severity in experimental PH models by mitigating pulmonary vascular remodeling.
Conclusions: A novel disulfide bond in cyclin D-CDK4 acts as a rapid switch to inhibit kinase activity and halt cell proliferation. This oxidative modification forms at a critical cysteine residue, which is unique to CDK4, offering the potential for the design of a selective covalent inhibitor predicted to be beneficial in PH.
Keywords: cell cycle; cell proliferation; hypertension, pulmonary; myocytes, smooth muscle; oxidation-reduction.
Conflict of interest statement
Figures

References
-
- Wilkins MR, Ghofrani HA, Weissmann N, Aldashev A, Zhao L. Pathophysiology and treatment of high-altitude pulmonary vascular disease. Circulation. 2015;131:582–590. doi: 10.1161/CIRCULATIONAHA.114.006977 - PubMed
-
- Masri FA, Xu W, Comhair SA, Asosingh K, Koo M, Vasanji A, Drazba J, Anand-Apte B, Erzurum SC. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;293:L548–L554. doi: 10.1152/ajplung.00428.2006 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
