

Predicting multiple conformations via sequence clustering and AlphaFold2
- PMID: 37956700
- PMCID: PMC10808063
- DOI: 10.1038/s41586-023-06832-9
Predicting multiple conformations via sequence clustering and AlphaFold2
Abstract
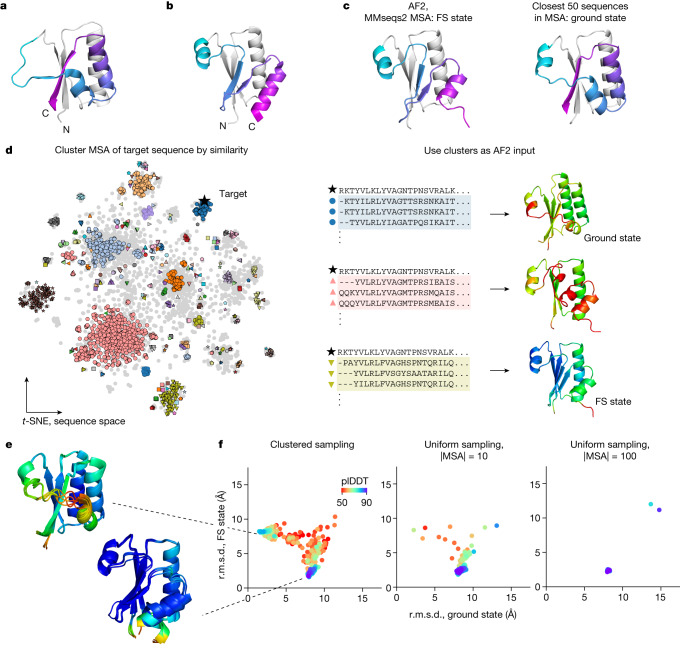
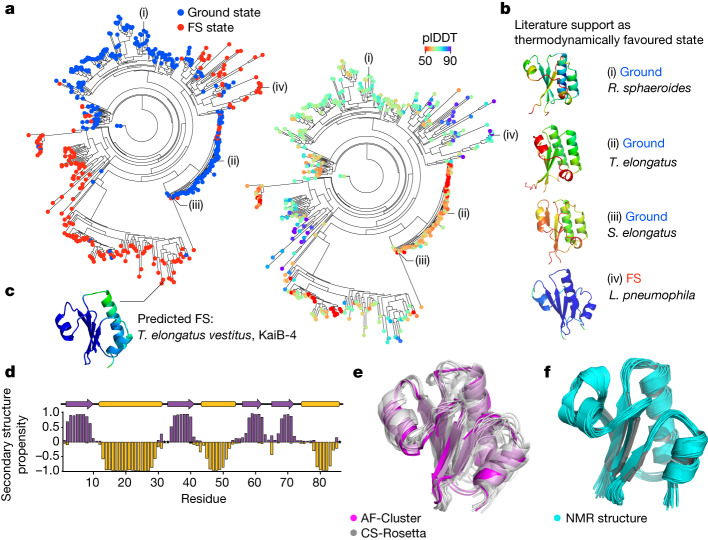
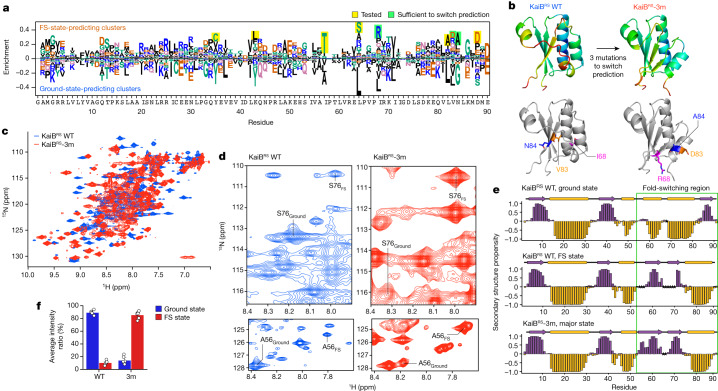
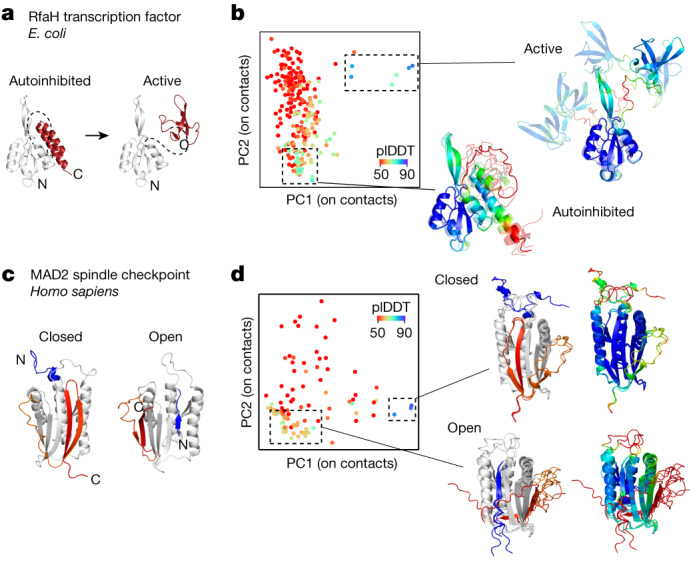
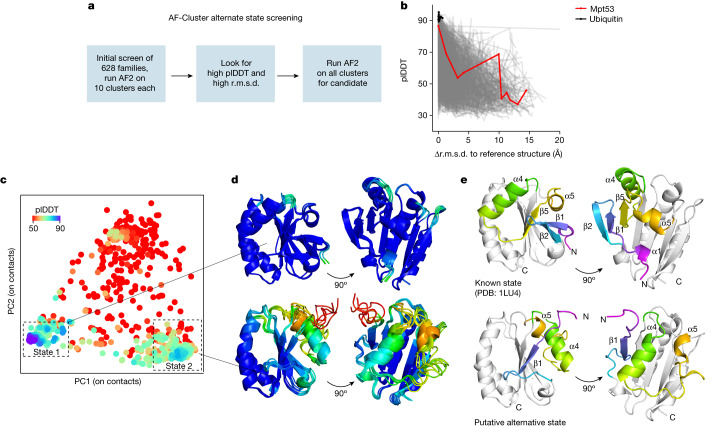
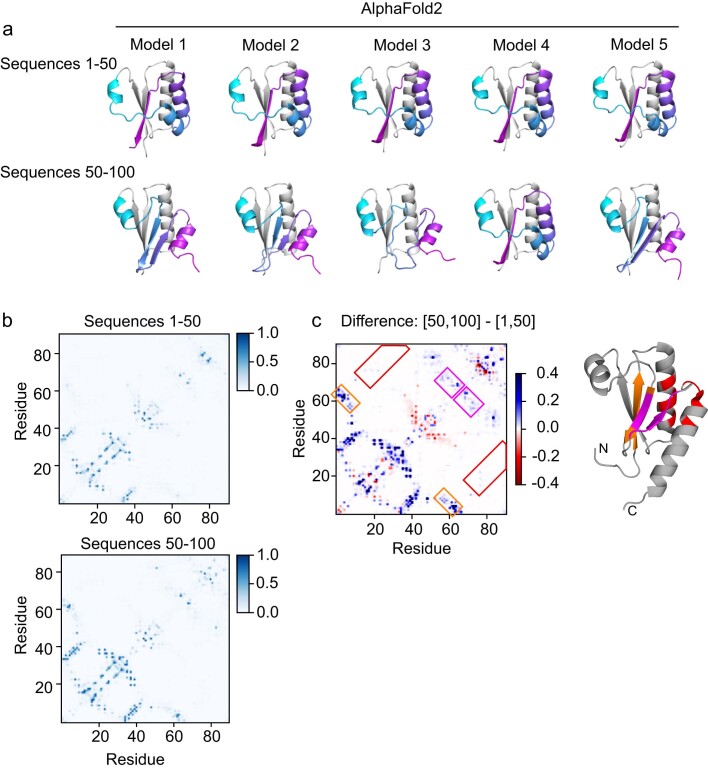
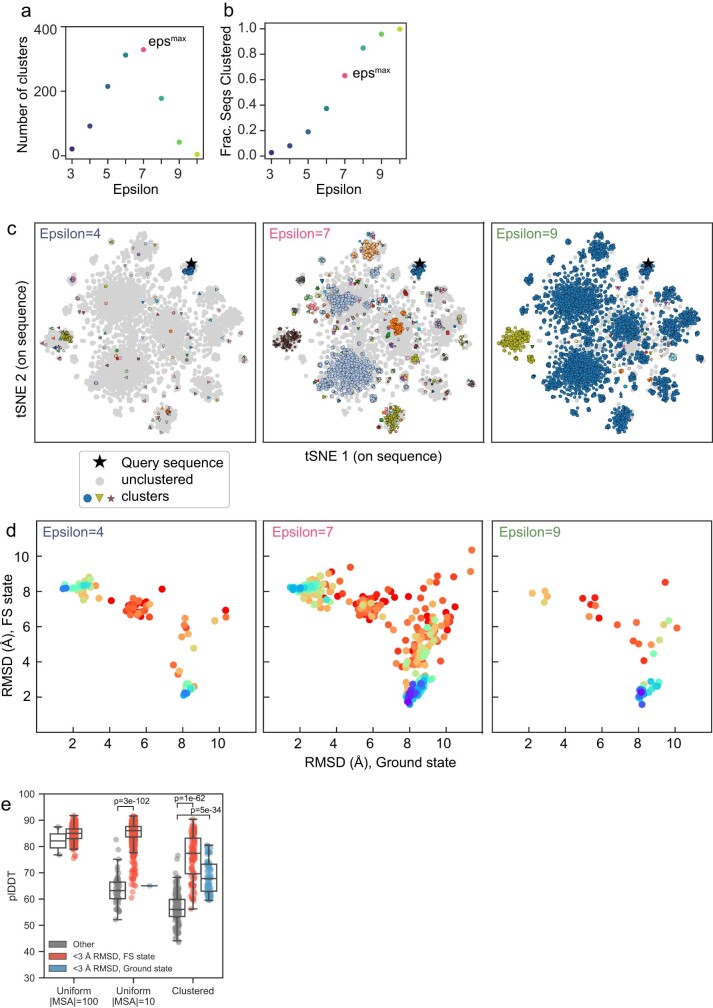
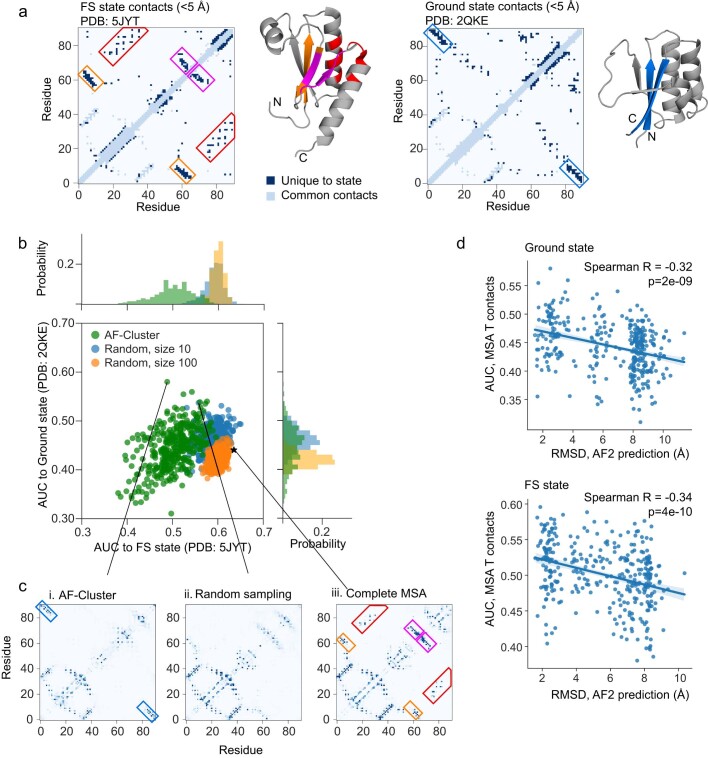
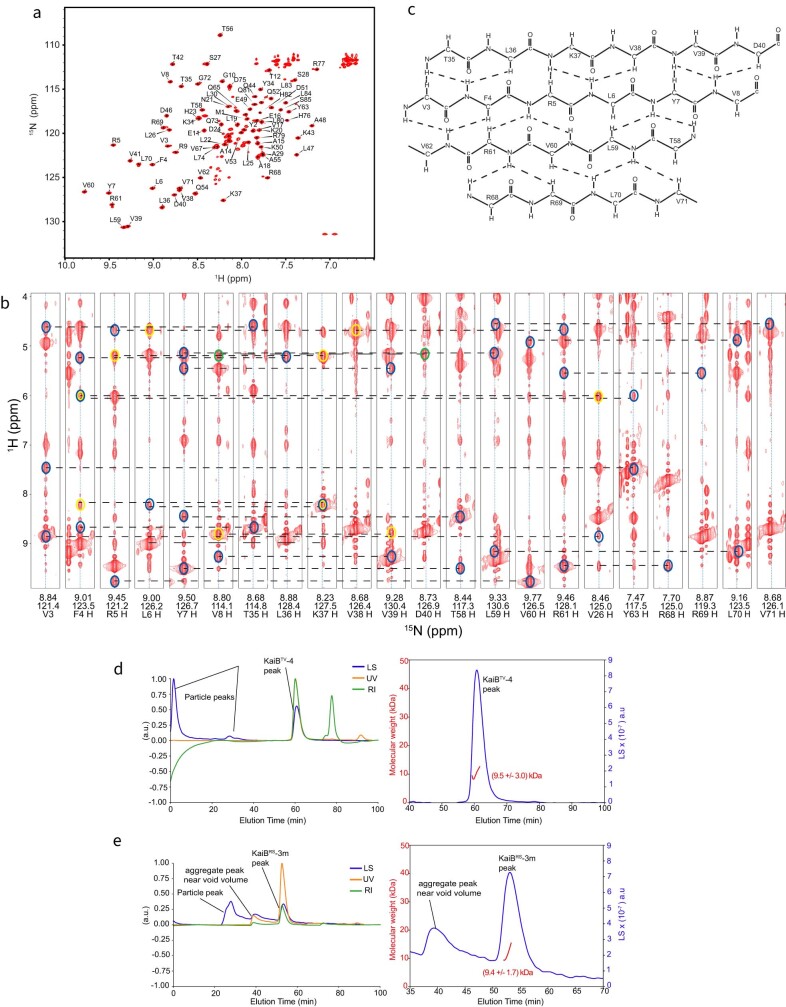
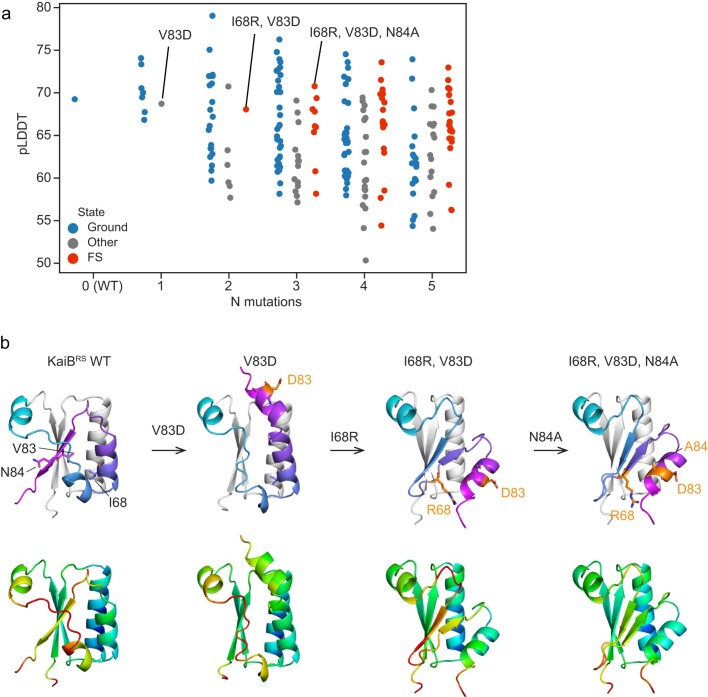
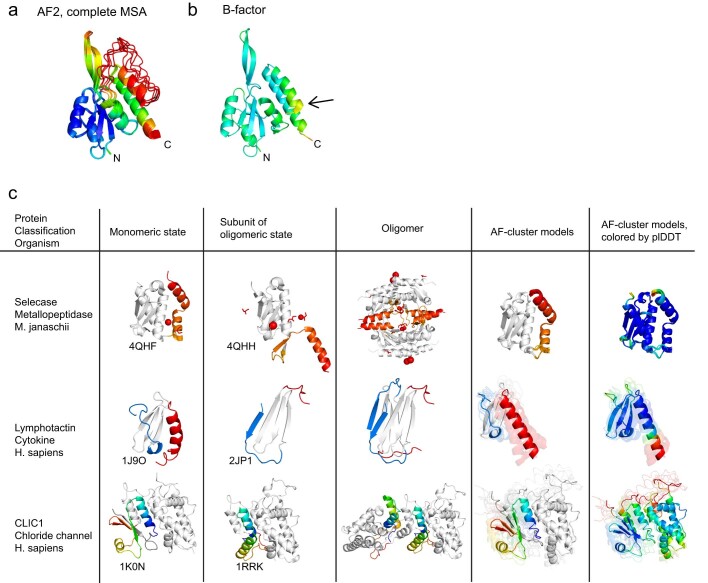
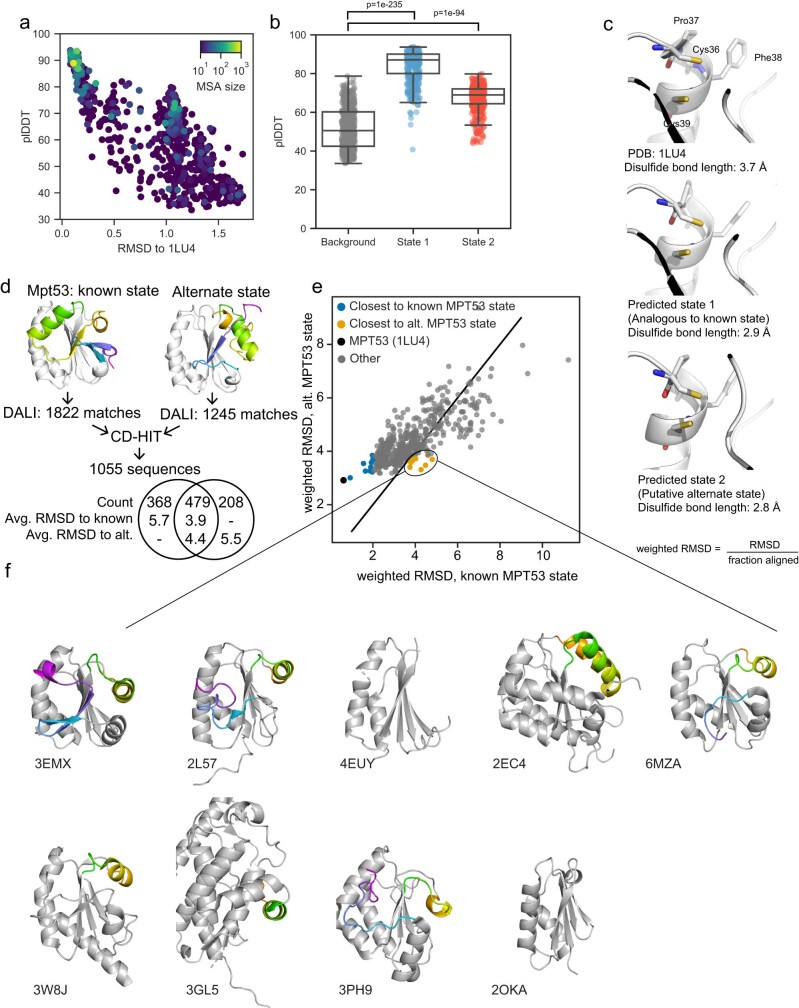
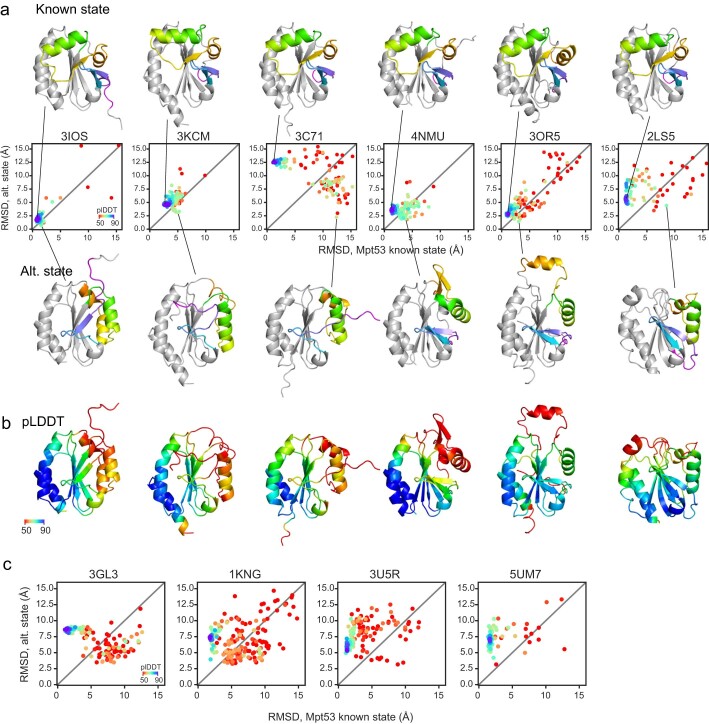
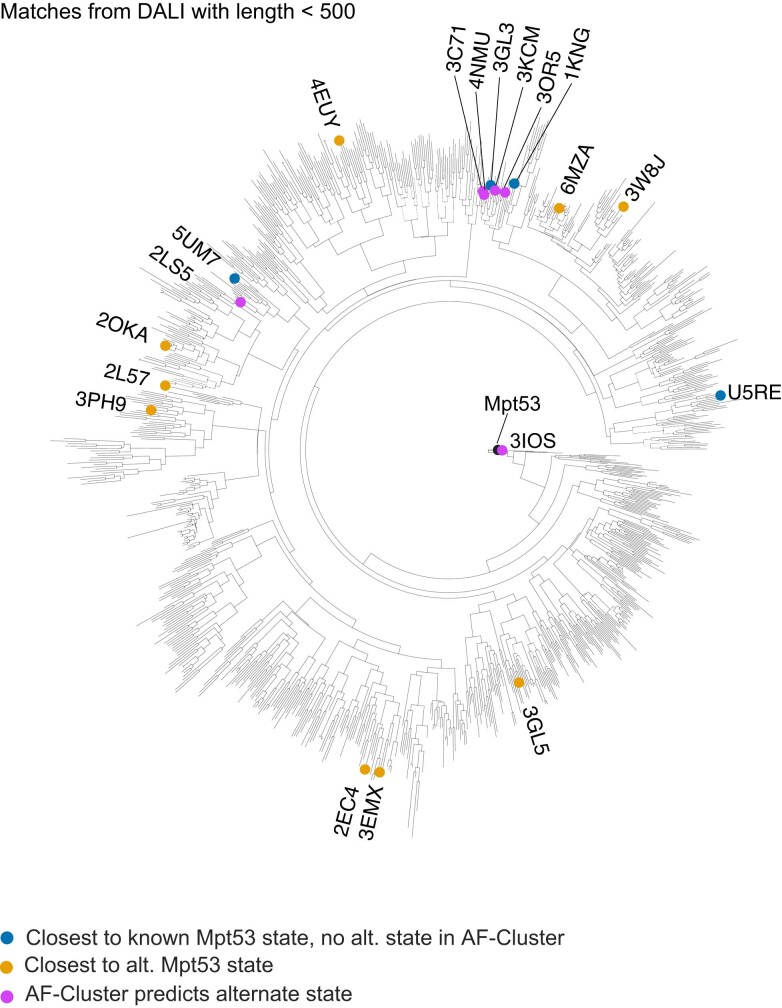
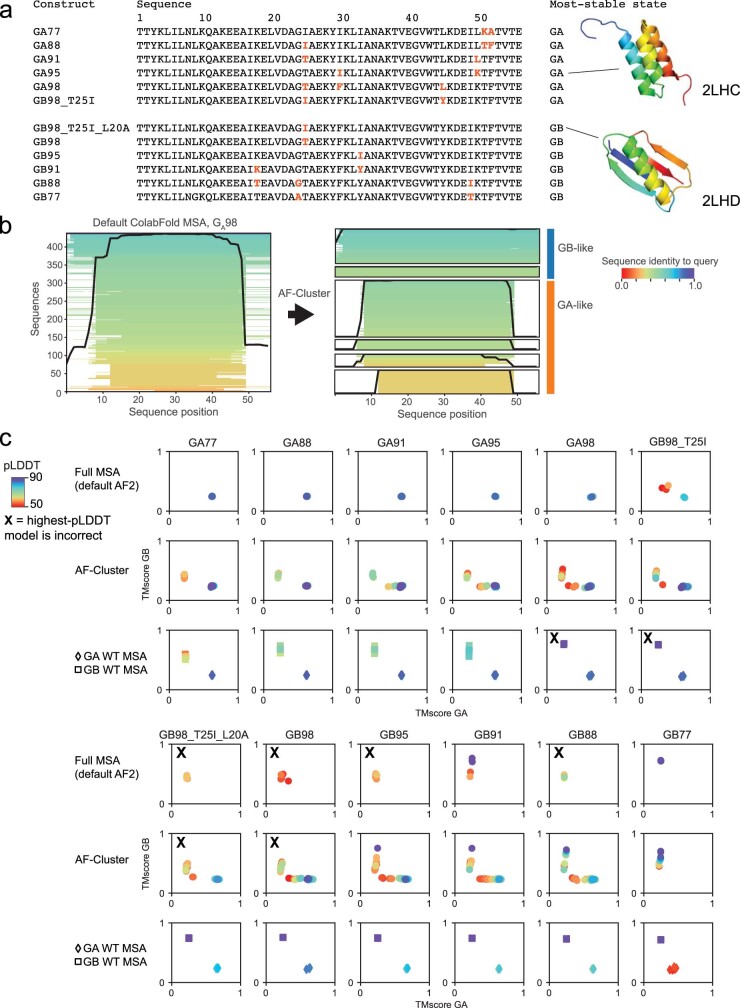
AlphaFold2 (ref. 1) has revolutionized structural biology by accurately predicting single structures of proteins. However, a protein's biological function often depends on multiple conformational substates2, and disease-causing point mutations often cause population changes within these substates3,4. We demonstrate that clustering a multiple-sequence alignment by sequence similarity enables AlphaFold2 to sample alternative states of known metamorphic proteins with high confidence. Using this method, named AF-Cluster, we investigated the evolutionary distribution of predicted structures for the metamorphic protein KaiB5 and found that predictions of both conformations were distributed in clusters across the KaiB family. We used nuclear magnetic resonance spectroscopy to confirm an AF-Cluster prediction: a cyanobacteria KaiB variant is stabilized in the opposite state compared with the more widely studied variant. To test AF-Cluster's sensitivity to point mutations, we designed and experimentally verified a set of three mutations predicted to flip KaiB from Rhodobacter sphaeroides from the ground to the fold-switched state. Finally, screening for alternative states in protein families without known fold switching identified a putative alternative state for the oxidoreductase Mpt53 in Mycobacterium tuberculosis. Further development of such bioinformatic methods in tandem with experiments will probably have a considerable impact on predicting protein energy landscapes, essential for illuminating biological function.
© 2023. The Author(s).
Conflict of interest statement
D.K. is a co-founder of Relay Therapeutics and MOMA Therapeutics. The other authors declare no competing interests.
Figures
Comment in
-
Sequence clustering confounds AlphaFold2.Nature. 2025 Feb;638(8051):E8-E12. doi: 10.1038/s41586-024-08267-2. Epub 2025 Feb 19. Nature. 2025. PMID: 39972235 No abstract available.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
