

This is a preprint.
A Germline Point Mutation in the MYC-FBW7 Phosphodegron Initiates Hematopoietic Malignancies
- PMID: 37961183
- PMCID: PMC10634767
- DOI: 10.1101/2023.10.23.563660
A Germline Point Mutation in the MYC-FBW7 Phosphodegron Initiates Hematopoietic Malignancies
Update in
-
A germline point mutation in the MYC-FBW7 phosphodegron initiates hematopoietic malignancies.Genes Dev. 2024 Apr 17;38(5-6):253-272. doi: 10.1101/gad.351292.123. Genes Dev. 2024. PMID: 38565249 Free PMC article.
Abstract
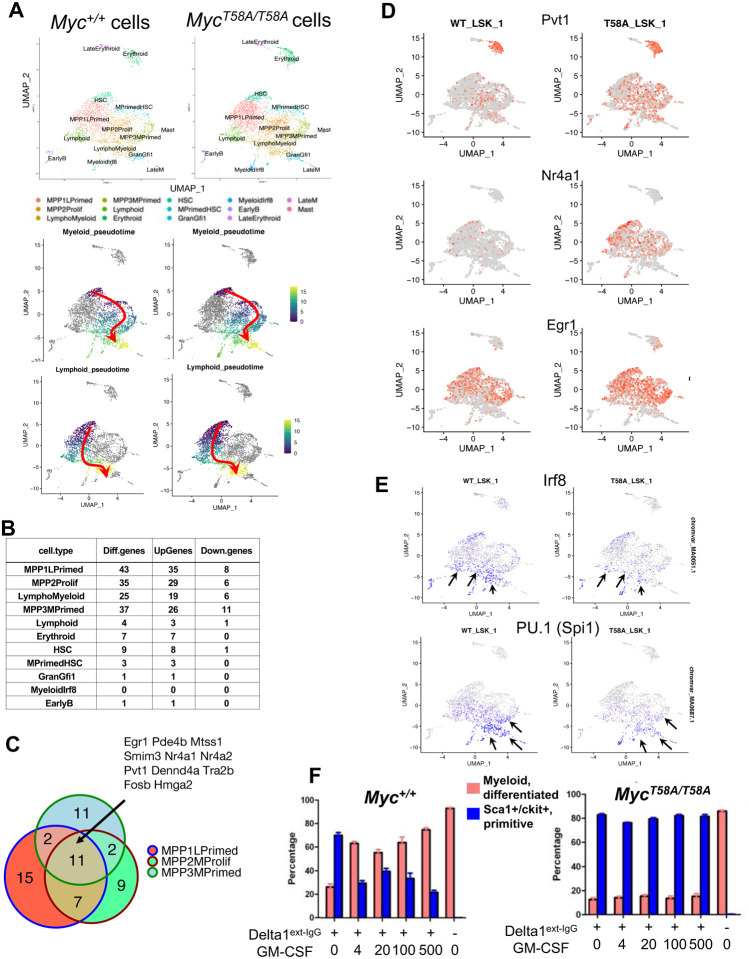
Oncogenic activation of MYC in cancers predominantly involves increased transcription rather than coding region mutations. However, MYC-dependent lymphomas frequently contain point mutations in the MYC phospho-degron, including at threonine-58 (T58), where phosphorylation permits binding by the FBW7 ubiquitin ligase triggering MYC degradation. To understand how T58 phosphorylation functions in normal cell physiology, we introduced an alanine mutation at T58 (T58A) into the endogenous c-Myc locus in the mouse germline. While MYC-T58A mice develop normally, lymphomas and myeloid leukemias emerge in ~60% of adult homozygous T58A mice. We find that primitive hematopoietic progenitor cells from MYC-T58A mice exhibit aberrant self-renewal normally associated with hematopoietic stem cells (HSCs) and upregulate a subset of Myc target genes important in maintaining stem/progenitor cell balance. Genomic occupancy by MYC-T58A was increased at all promoters, compared to WT MYC, while genes differentially expressed in a T58A-dependent manner were significantly more proximal to MYC-bound enhancers. MYC-T58A lymphocyte progenitors exhibited metabolic alterations and decreased activation of inflammatory and apoptotic pathways. Our data demonstrate that a single point mutation in Myc is sufficient to produce a profound gain of function in multipotential hematopoietic progenitors associated with self-renewal and initiation of lymphomas and leukemias.
Conflict of interest statement
COMPETING INTEREST STATEMENT Robert N Eisenman: Scientific Advisory Board Member: Kronos Bio Inc.; Shenogen Pharma Beijing. No overlap with the present study. The other authors declare that no competing interests exist.
Figures





References
-
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. 1985. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 318: 533–538. - PubMed
-
- Babcock JT, Nguyen HB, He Y, Hendricks JW, Wek RC, Quilliam LA. 2013. Mammalian target of rapamycin complex 1 (mTORC1) enhances bortezomib-induced death in tuberous sclerosis complex (TSC)-null cells by a c-MYC-dependent induction of the unfolded protein response. J Biol Chem 288: 15687–15698. - PMC - PubMed
-
- Bhatia K, Huppi K, Spangler G, Siwarski D, Iyer R, Magrath I. 1993. Point mutations in the c-Myc transactivation domain are common in Burkid’s lymphoma and mouse plasmacytomas. Nat Genet 5: 56–61. - PubMed
-
- Bhatia K, Spangler G, Gaidano G, Hamdy N, Dalla-Favera R, Magrath I. 1994. Mutations in the coding region of c-myc occur frequently in acquired immunodeficiency syndrome-associated lymphomas. Blood 84: 883–888. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases