

This is a preprint.
Decoding Heterogenous Single-cell Perturbation Responses
- PMID: 37961332
- PMCID: PMC10635009
- DOI: 10.1101/2023.10.30.564796
Decoding Heterogenous Single-cell Perturbation Responses
Update in
-
Decoding heterogeneous single-cell perturbation responses.Nat Cell Biol. 2025 Mar;27(3):493-504. doi: 10.1038/s41556-025-01626-9. Epub 2025 Feb 26. Nat Cell Biol. 2025. PMID: 40011559 Free PMC article.
Abstract
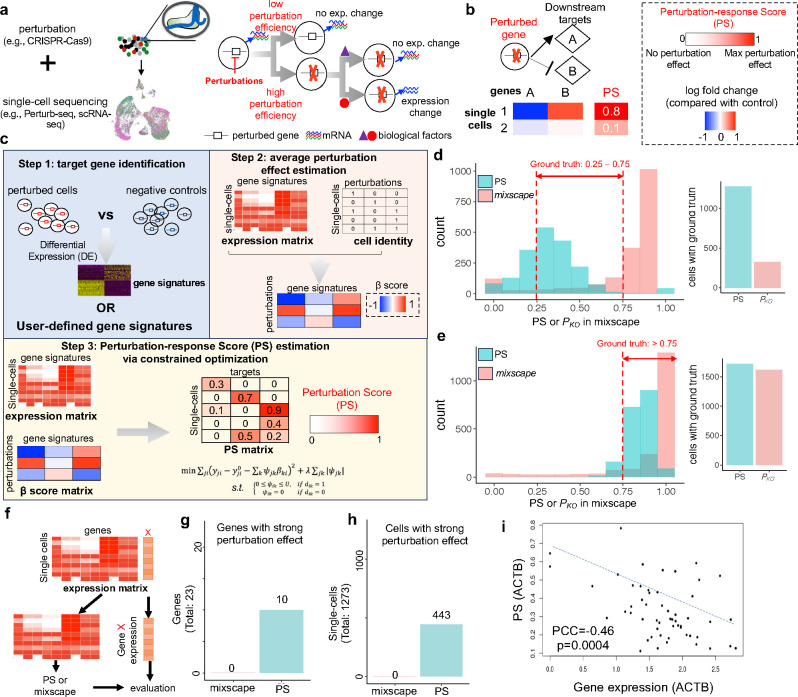
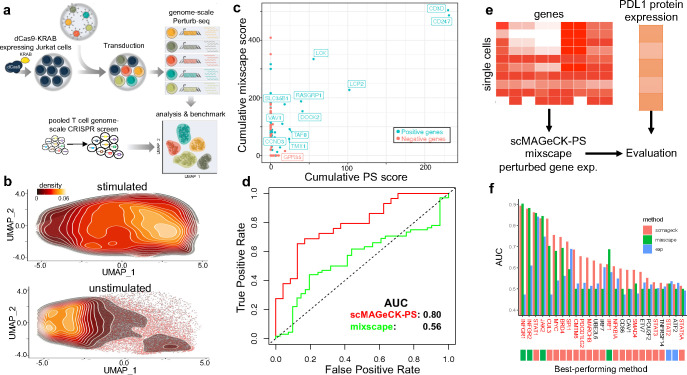
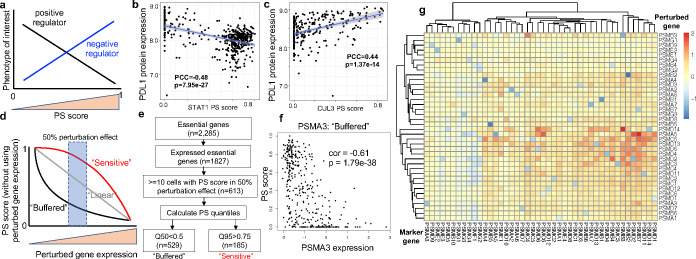
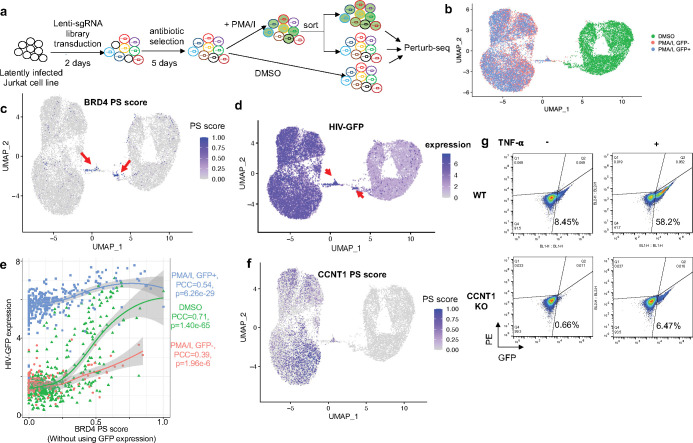
Understanding diverse responses of individual cells to the same perturbation is central to many biological and biomedical problems. Current methods, however, do not precisely quantify the strength of perturbation responses and, more importantly, reveal new biological insights from heterogeneity in responses. Here we introduce the perturbation-response score (PS), based on constrained quadratic optimization, to quantify diverse perturbation responses at a single-cell level. Applied to single-cell transcriptomes of large-scale genetic perturbation datasets (e.g., Perturb-seq), PS outperforms existing methods for quantifying partial gene perturbation responses. In addition, PS presents two major advances. First, PS enables large-scale, single-cell-resolution dosage analysis of perturbation, without the need to titrate perturbation strength. By analyzing the dose-response patterns of over 2,000 essential genes in Perturb-seq, we identify two distinct patterns, depending on whether a moderate reduction in their expression induces strong downstream expression alterations. Second, PS identifies intrinsic and extrinsic biological determinants of perturbation responses. We demonstrate the application of PS in contexts such as T cell stimulation, latent HIV-1 expression, and pancreatic cell differentiation. Notably, PS unveiled a previously unrecognized, cell-type-specific role of coiled-coil domain containing 6 (CCDC6) in guiding liver and pancreatic lineage decisions, where CCDC6 knockouts drive the endoderm cell differentiation towards liver lineage, rather than pancreatic lineage. The PS approach provides an innovative method for dose-to-function analysis and will enable new biological discoveries from single-cell perturbation datasets.
Keywords: CRISPR-based genetic perturbations; Perturb-seq; computational model; single-cell RNA-seq.
Conflict of interest statement
Competing interests T.B. is a co-founder and Managing Director of Myllia Biotechnology. A.K., A.V., N.U. and A.L. are employees of Myllia Biotechnology. Other authors declare that they have no competing interest.
Figures
References
-
- Jaitin DA, Weiner A, Yofe I, Lara-Astiaso D, Keren-Shaul H, David E, et al. Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq. Cell 2016;167:1883–1896.e15. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases