

This is a preprint.
High-resolution CTCF footprinting reveals impact of chromatin state on cohesin extrusion dynamics
- PMID: 37961446
- PMCID: PMC10634716
- DOI: 10.1101/2023.10.20.563340
High-resolution CTCF footprinting reveals impact of chromatin state on cohesin extrusion dynamics
Update in
-
High-resolution CTCF footprinting reveals impact of chromatin state on cohesin extrusion.Nat Commun. 2025 May 15;16(1):4506. doi: 10.1038/s41467-025-57775-w. Nat Commun. 2025. PMID: 40374602 Free PMC article.
Abstract
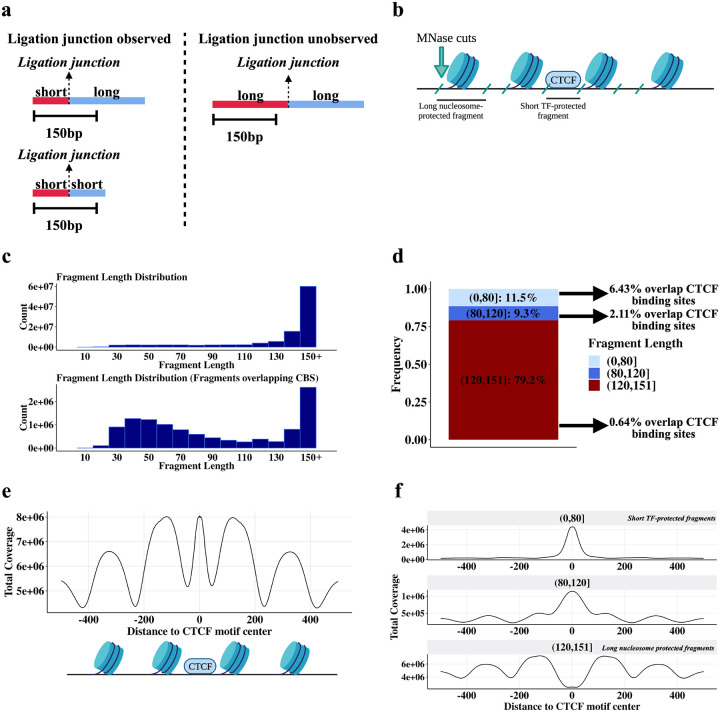
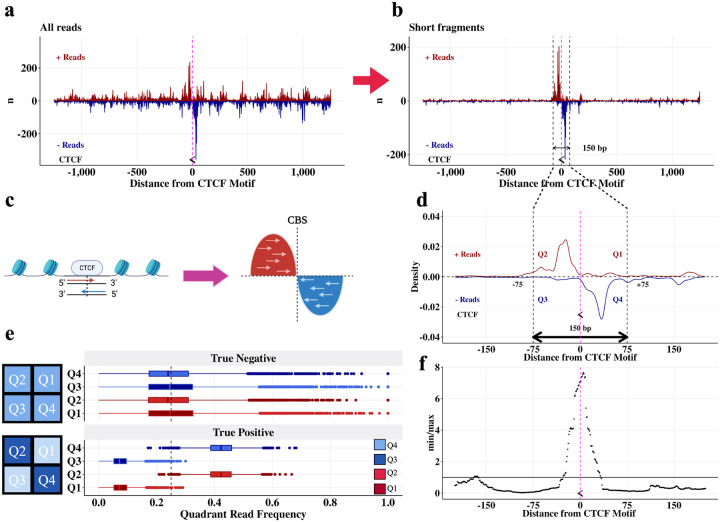
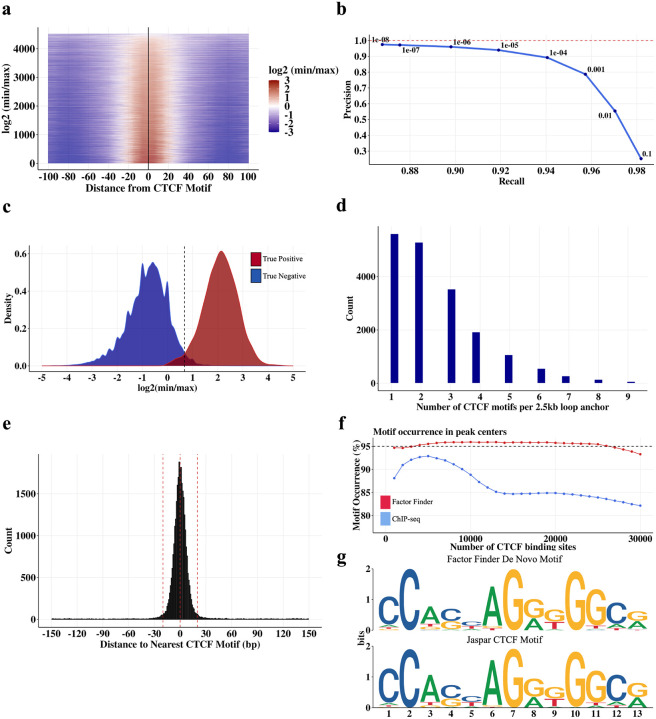
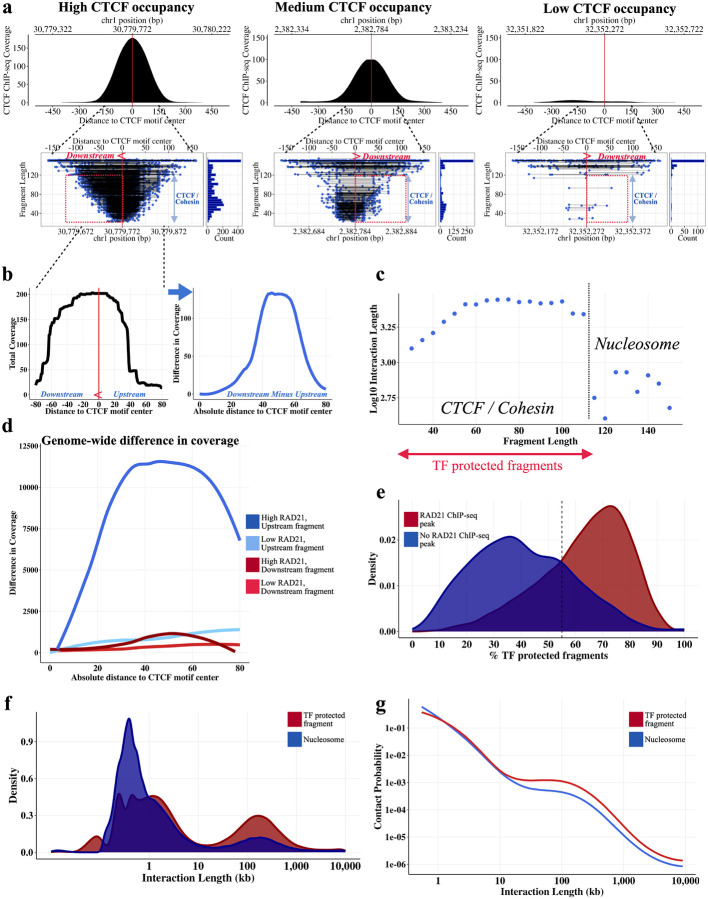
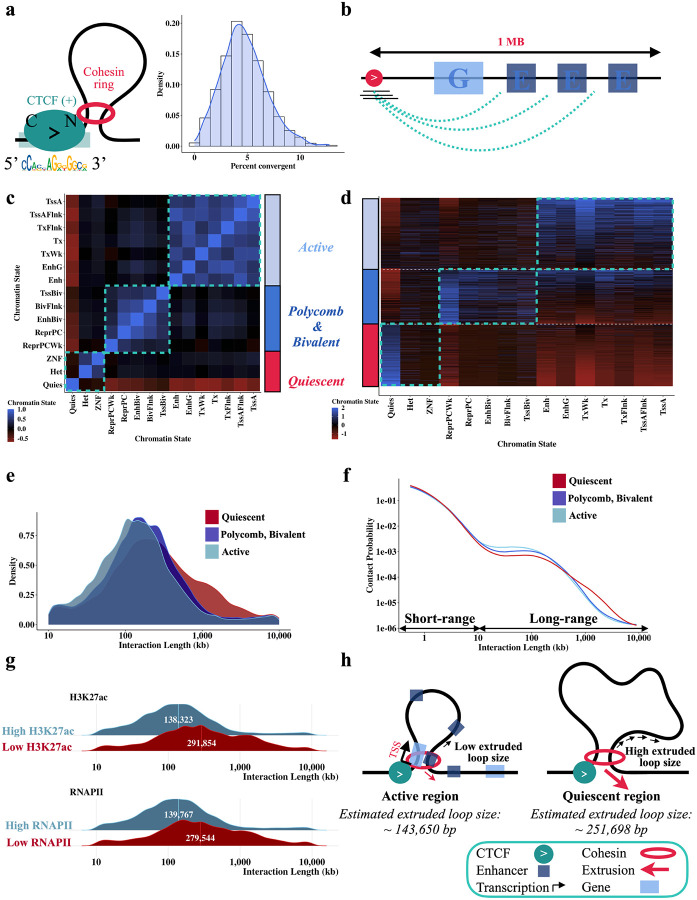
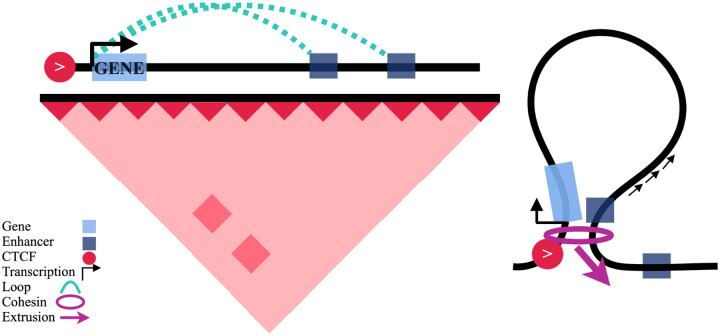
DNA looping is vital for establishing many enhancer-promoter interactions. While CTCF is known to anchor many cohesin-mediated loops, the looped chromatin fiber appears to predominantly exist in a poorly characterized actively extruding state. To better characterize extruding chromatin loop structures, we used CTCF MNase HiChIP data to determine both CTCF binding at high resolution and 3D contact information. Here we present FactorFinder, a tool that identifies CTCF binding sites at near base-pair resolution. We leverage this substantial advance in resolution to determine that the fully extruded (CTCF-CTCF) state is rare genome-wide with locus-specific variation from ~1-10%. We further investigate the impact of chromatin state on loop extrusion dynamics, and find that active enhancers and RNA Pol II impede cohesin extrusion, facilitating an enrichment of enhancer-promoter contacts in the partially extruded loop state. We propose a model of topological regulation whereby the transient, partially extruded states play active roles in transcription.
Conflict of interest statement
Disclosures Dovetail Genomics/Cantata Bio provided reagents and sample processing for HiChIP experiments. M.B. and M.S.B were employees at Dovetail Genomics during the course of this research. M.J.A has financial and consulting interests unrelated to this work in SeQure Dx and Chroma Medicine. M.J.A’s interests are reviewed and managed by Dana Farber Cancer Institute. J.K.J. is a co-founder of and has a financial interest in SeQure, Dx, Inc., a company developing technologies for gene editing target profiling. JKJ also has, or had during the course of this research, financial interests in several companies developing gene editing technology: Beam Therapeutics, Blink Therapeutics, Chroma Medicine, Editas Medicine, EpiLogic Therapeutics, Excelsior Genomics, Hera Biolabs, Monitor Biotechnologies, Nvelop Therapeutics (f/k/a ETx, Inc.), Pairwise Plants, Poseida Therapeutics, and Verve Therapeutics. J.K.J.’s interests were reviewed and are managed by Massachusetts General Hospital and Mass General Brigham in accordance with their conflict of interest policies.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources