

This is a preprint.
Dissecting signaling regulators driving AXL-mediated bypass resistance and associated phenotypes by phosphosite perturbations
- PMID: 37961516
- PMCID: PMC10634689
- DOI: 10.1101/2023.10.20.563266
Dissecting signaling regulators driving AXL-mediated bypass resistance and associated phenotypes by phosphosite perturbations
Abstract
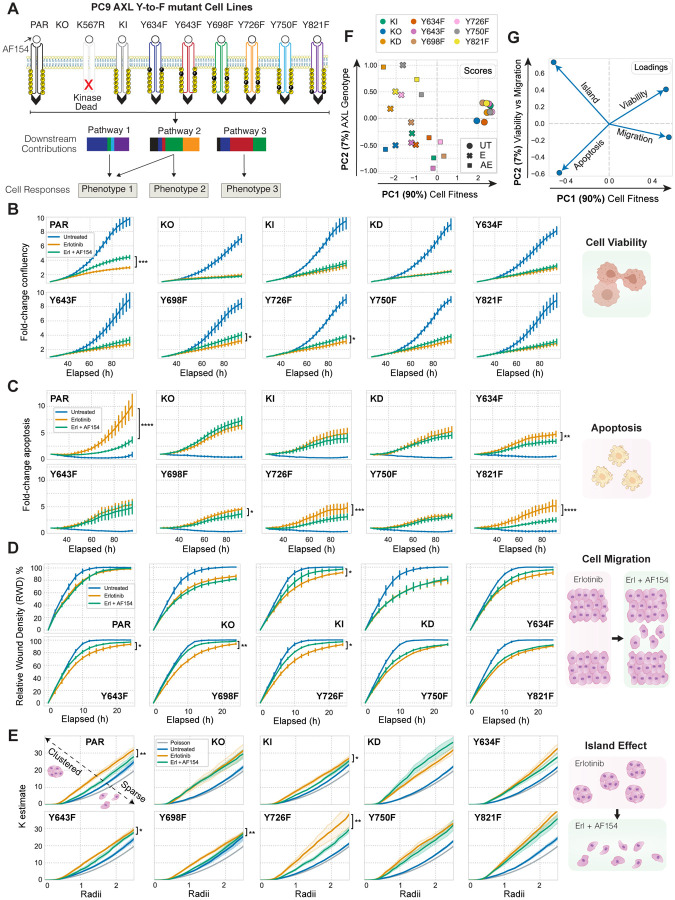
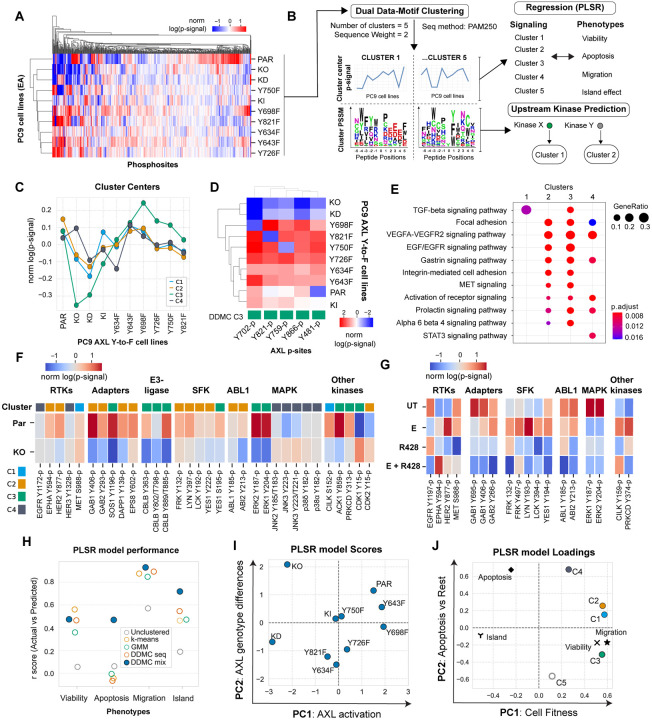
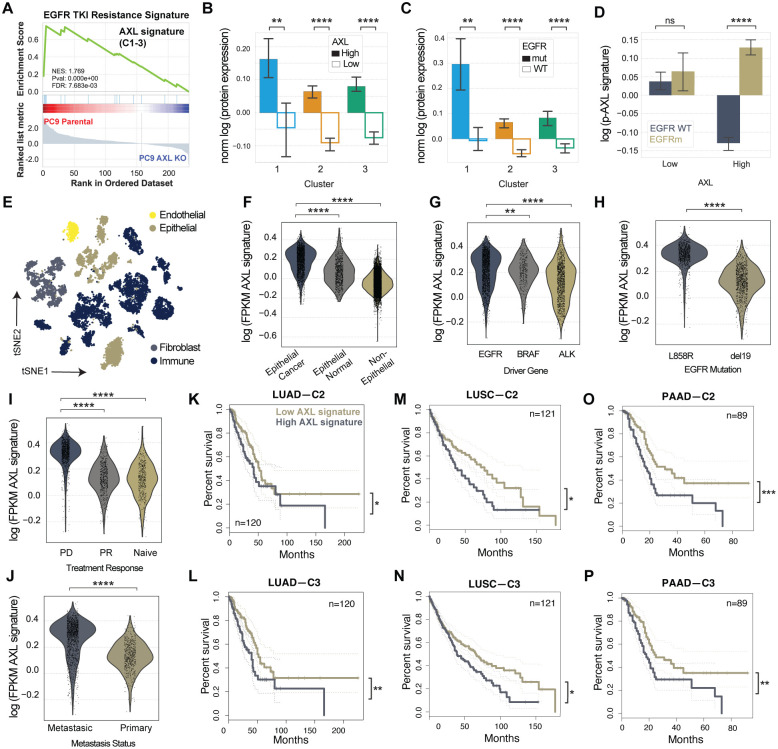
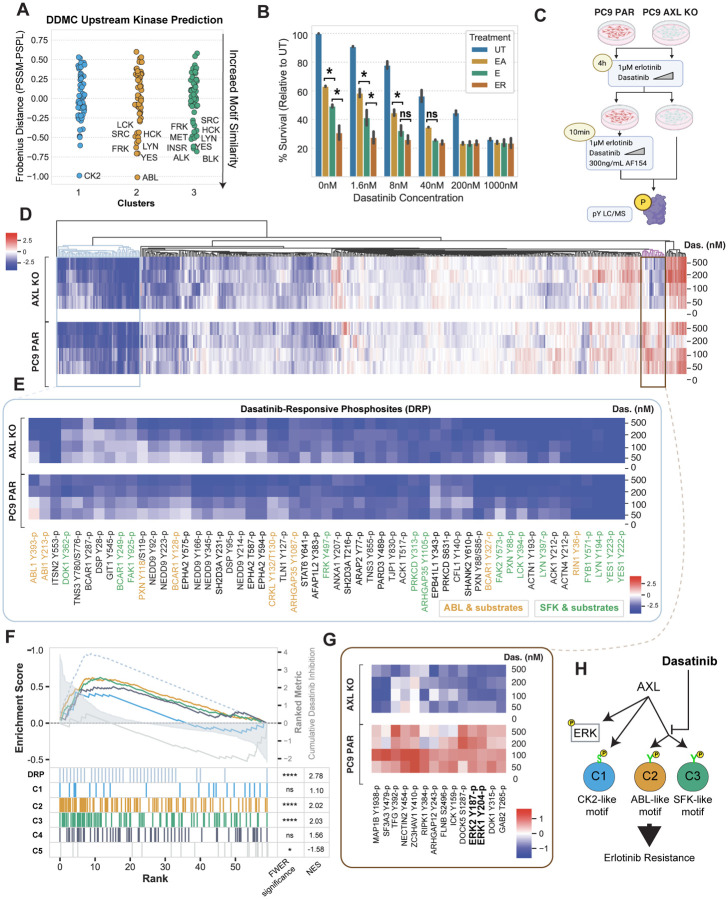
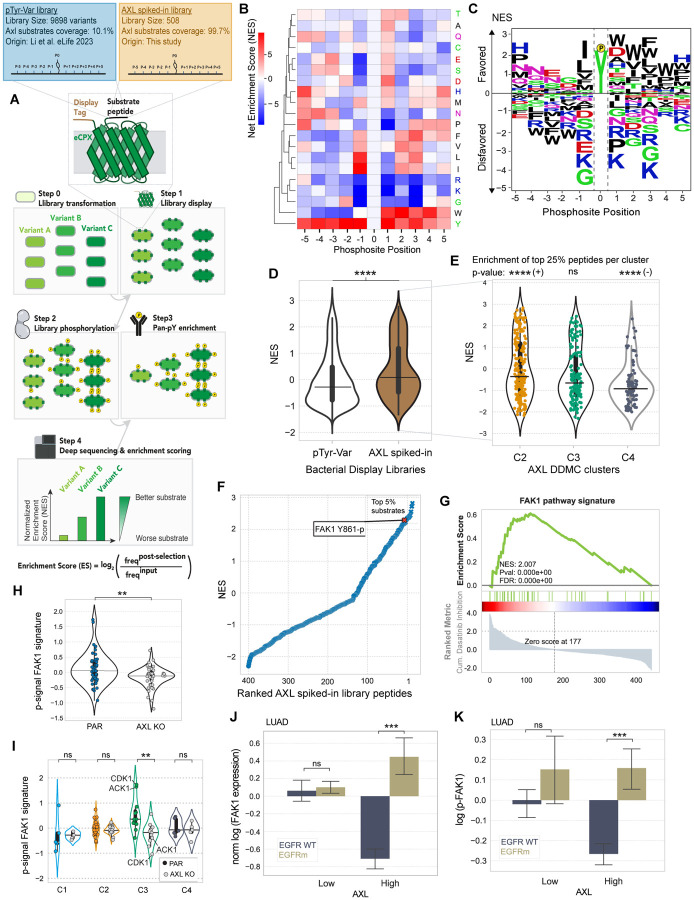
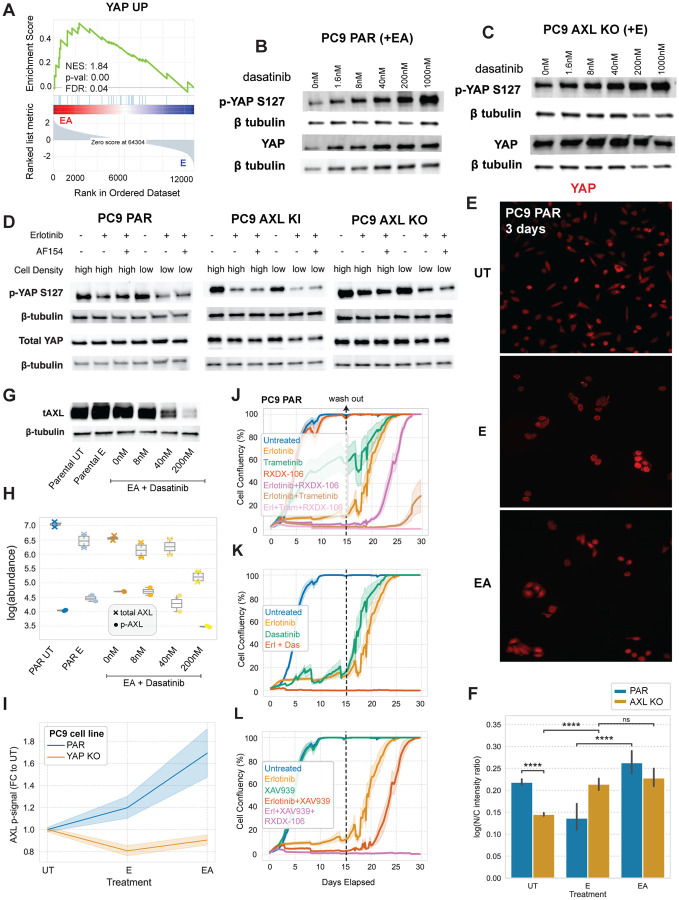
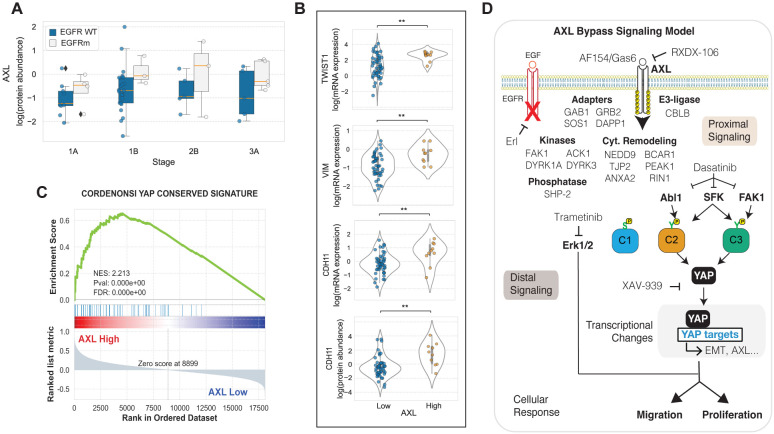
Receptor tyrosine kinase (RTK)-targeted therapies are often effective but invariably limited by drug resistance. A major mechanism of acquired resistance involves "bypass" switching to alternative pathways driven by non-targeted RTKs that restore proliferation. One such RTK is AXL whose overexpression, frequently observed in bypass resistant tumors, drives both cell survival and associated malignant phenotypes such as epithelial-to-mesenchymal (EMT) transition and migration. However, the signaling molecules and pathways eliciting these responses have remained elusive. To explore these coordinated effects, we generated a panel of mutant lung adenocarcinoma PC9 cell lines in which each AXL intracellular tyrosine residue was mutated to phenylalanine. By integrating measurements of phosphorylation signaling and other phenotypic changes associated with resistance through multivariate modeling, we mapped signaling perturbations to specific resistant phenotypes. Our results suggest that AXL signaling can be summarized into two clusters associated with progressive disease and poor clinical outcomes in lung cancer patients. These clusters displayed favorable Abl1 and SFK motifs and their phosphorylation was consistently decreased by dasatinib. High-throughput kinase specificity profiling showed that AXL likely activates the SFK cluster through FAK1 which is known to complex with Src. Moreover, the SFK cluster overlapped with a previously established focal adhesion kinase (FAK1) signature conferring EMT-mediated erlotinib resistance in lung cancer cells. Finally, we show that downstream of this kinase signaling, AXL and YAP form a positive feedback loop that sustains drug tolerant persister cells. Altogether, this work demonstrates an approach for dissecting signaling regulators by which AXL drives erlotinib resistance-associated phenotypic changes.
Conflict of interest statement
Competing interests: The authors declare that they have no competing interests.
Figures
References
-
- Atlanta G., Cancer Facts & Figures 2022. American Cancer Society (2022).
-
- Hospital X., Hu C., Hospital S. X., Zhou C., Wu Y.-L., Chen G., Feng J., Liu X.-Q., Wang C., Zhang S., Wang J., Zhou S., Ren S., Lu S., Zhang L., Hu C., Luo Y., Chen L., Ye M., Huang J., Zhi X., Zhang Y., Xiu Q., Ma J., You C., Erlotinib versus chemotherapy as firstline treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. The Lancet. 12, 735–777 (2011). - PubMed
-
- Ramalingam S. S., Vansteenkiste J., Planchard D., Cho B. C., Gray J. E., Ohe Y., Zhou C., Reungwetwattana T., Cheng Y., Chewaskulyong B., Shah R., Cobo M., Lee K. H., Cheema P., Tiseo M., John T., Lin M.-C., Imamura F., Kurata T., Todd A., Hodge R., Saggese M., Rukazenkov Y., Soria J.-C., Overall Survival with Osimertinib in Untreated, EGFR -Mutated Advanced NSCLC. New England Journal of Medicine. 382, 41–50 (2020). - PubMed
-
- Wilson T. R., Fridlyand J., Yan Y., Penuel E., Burton L., Chan E., Peng J., Lin E., Wang Y., Sosman J., Ribas A., Li J., Moffat J., Sutherlin D. P., Koeppen H., Merchant M., Neve R., Settleman J., Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 487, 505–509 (2012). - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous