

This is a preprint.
Programs, Origins, and Niches of Immunomodulatory Myeloid Cells in Gliomas
- PMID: 37961527
- PMCID: PMC10634776
- DOI: 10.1101/2023.10.24.563466
Programs, Origins, and Niches of Immunomodulatory Myeloid Cells in Gliomas
Update in
-
Programs, origins and immunomodulatory functions of myeloid cells in glioma.Nature. 2025 Apr;640(8060):1072-1082. doi: 10.1038/s41586-025-08633-8. Epub 2025 Feb 26. Nature. 2025. PMID: 40011771 Free PMC article.
Abstract
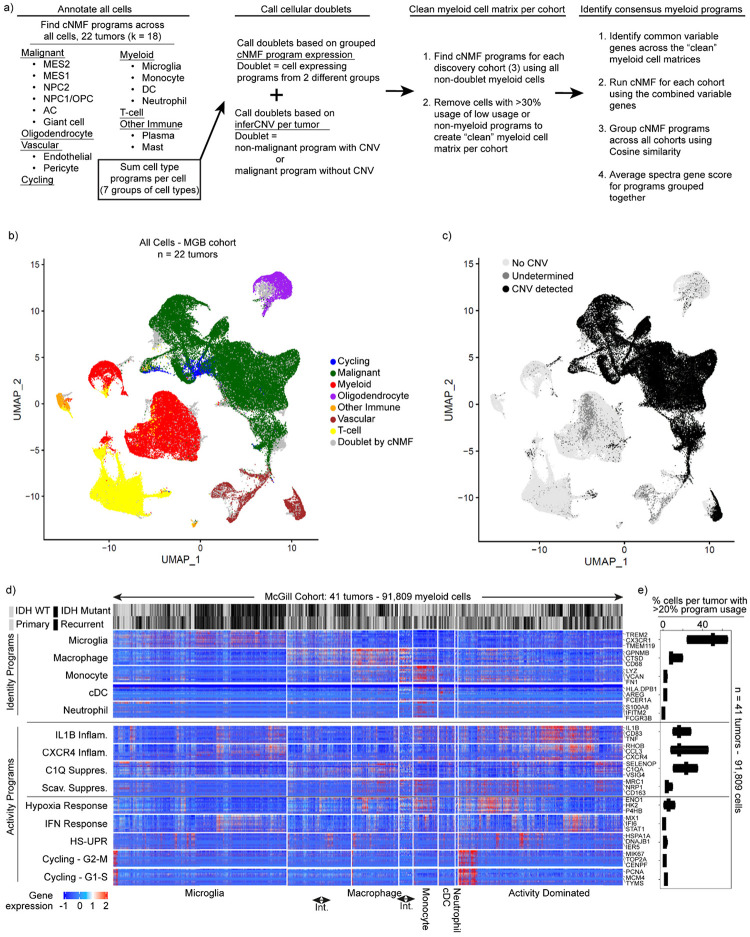
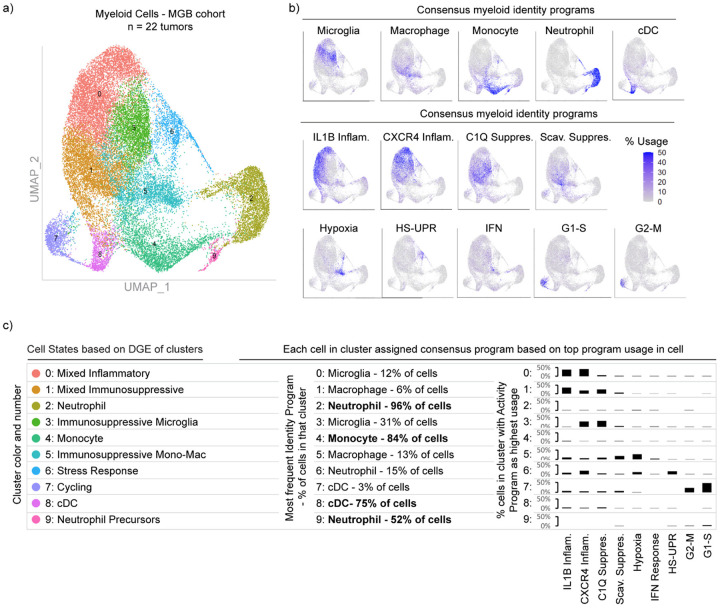
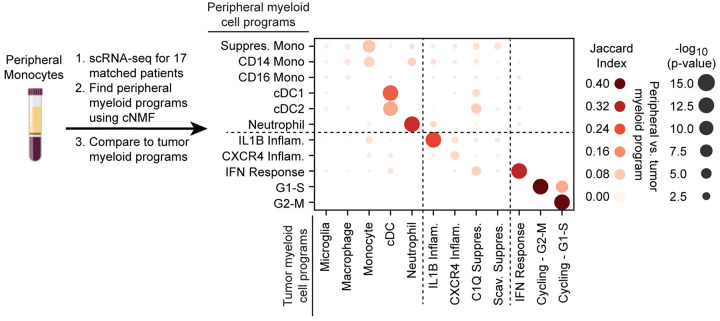
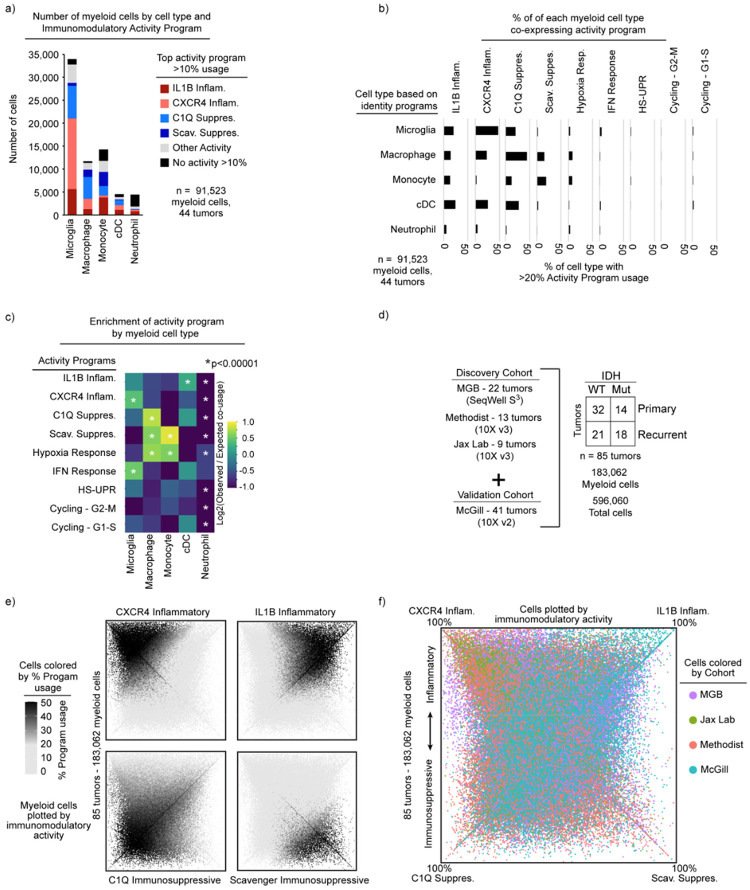
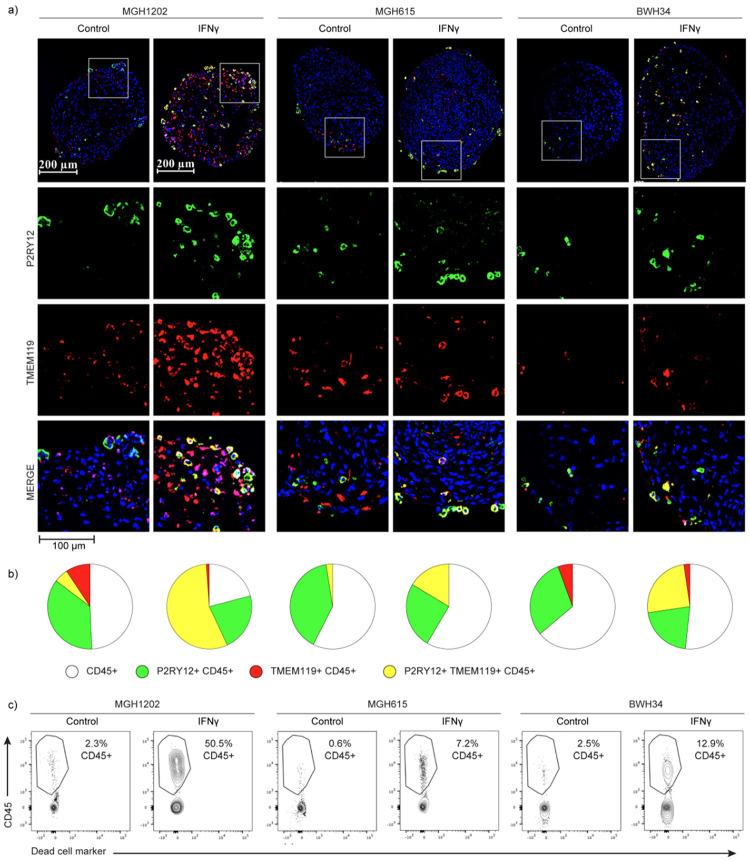
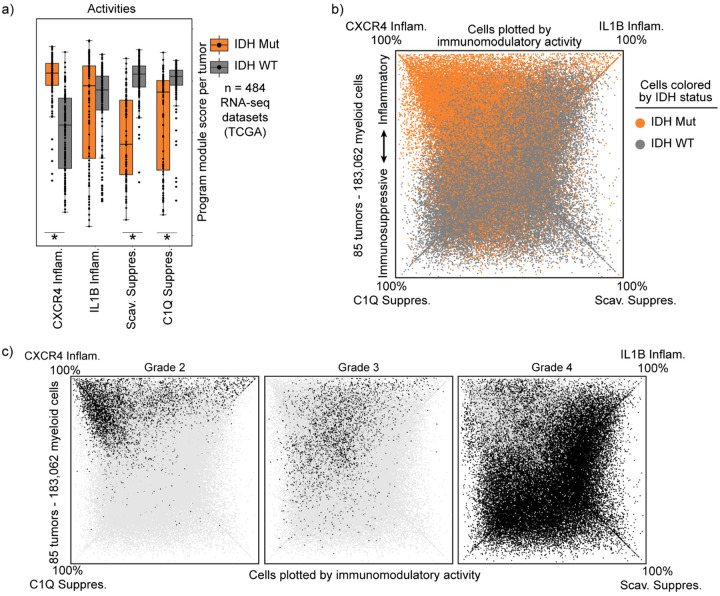
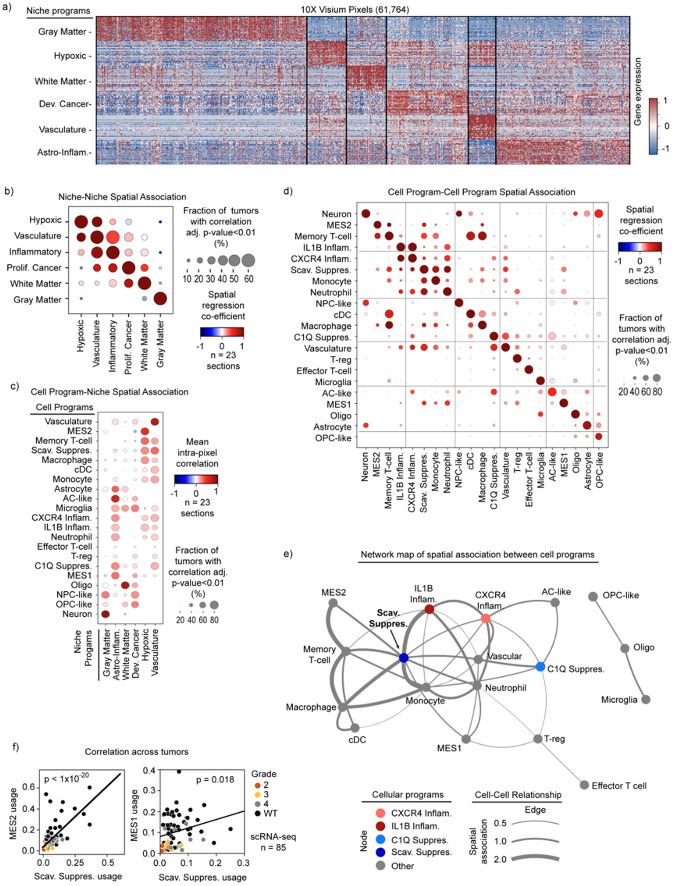
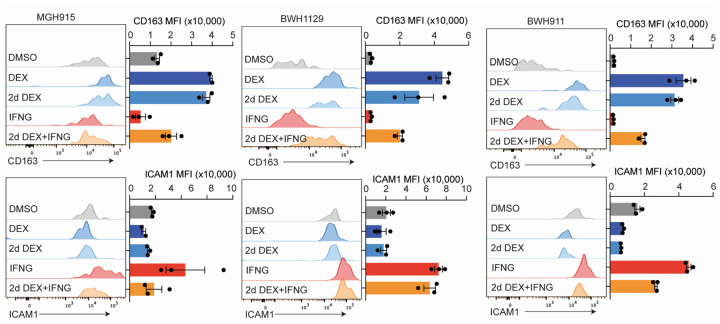
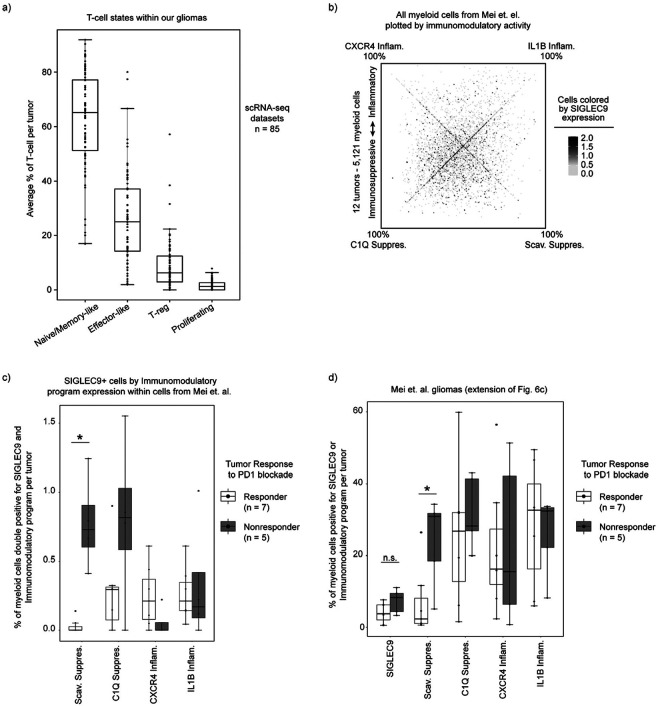
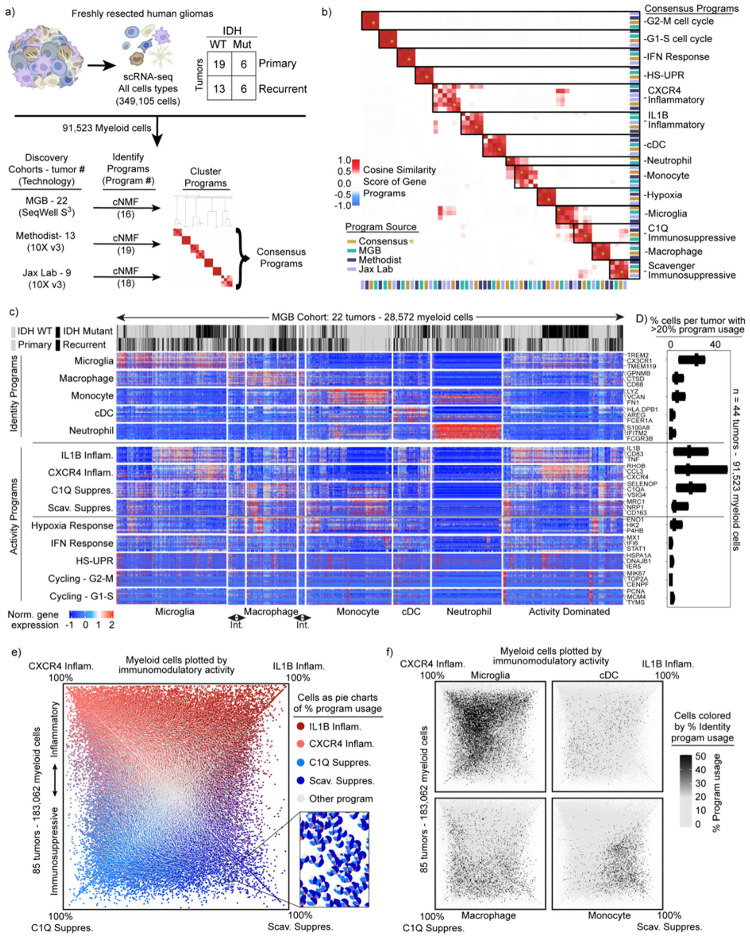
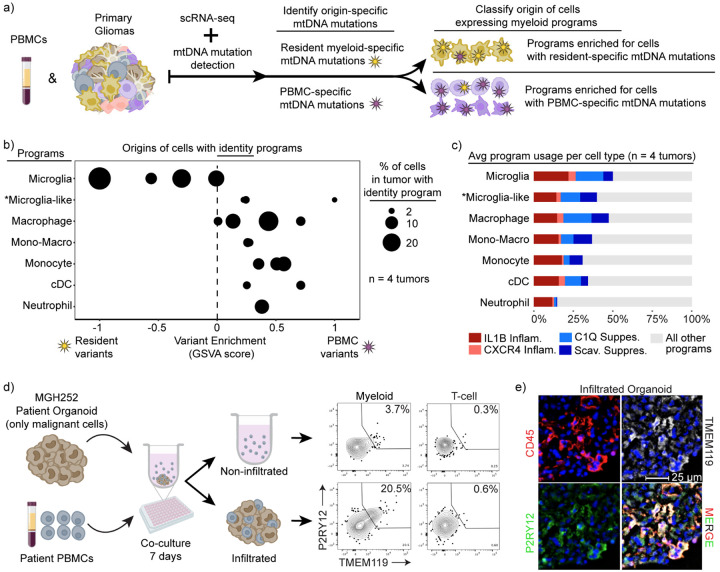
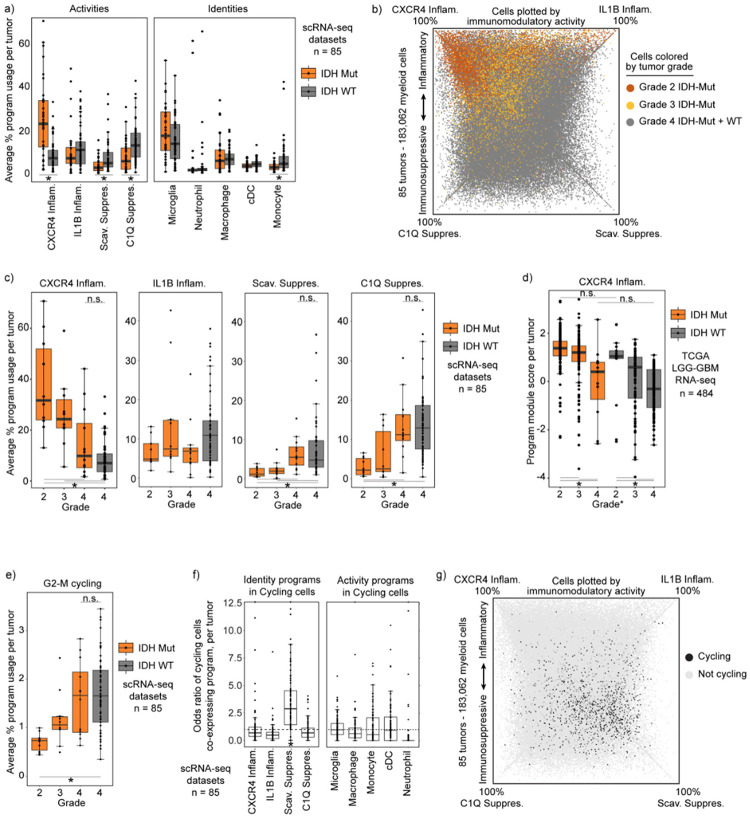
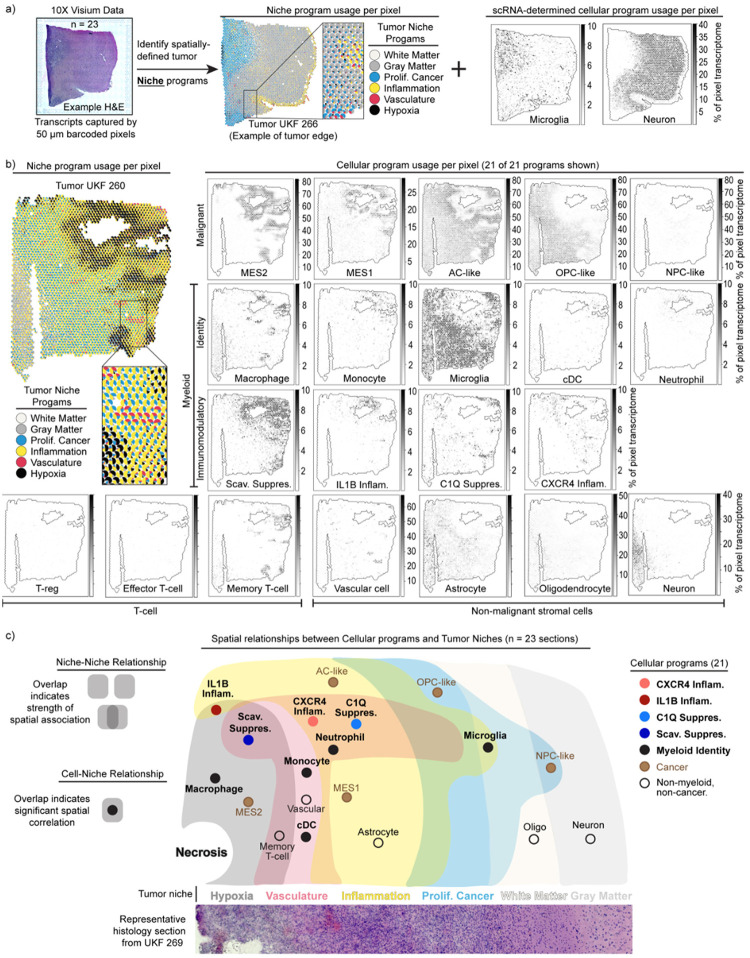
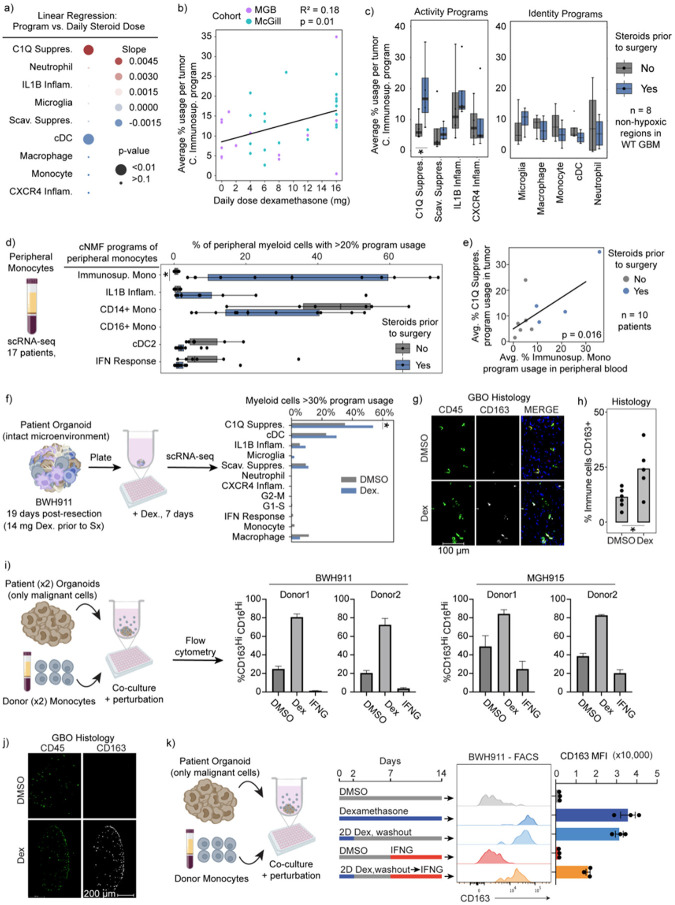
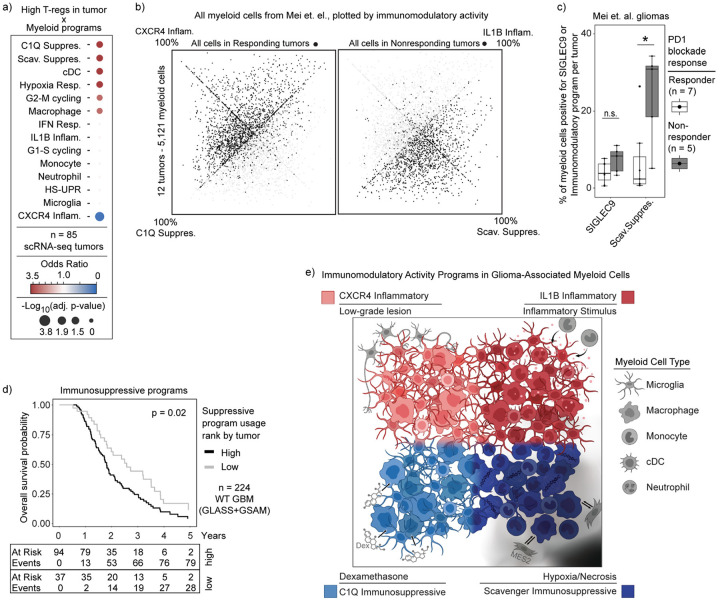
Gliomas are incurable malignancies notable for an immunosuppressive microenvironment with abundant myeloid cells whose immunomodulatory properties remain poorly defined. Here, utilizing scRNA-seq data for 183,062 myeloid cells from 85 human tumors, we discover that nearly all glioma-associated myeloid cells express at least one of four immunomodulatory activity programs: Scavenger Immunosuppressive, C1Q Immunosuppressive, CXCR4 Inflammatory, and IL1B Inflammatory. All four programs are present in IDH1 mutant and wild-type gliomas and are expressed in macrophages, monocytes, and microglia whether of blood or resident myeloid cell origins. Integrating our scRNA-seq data with mitochondrial DNA-based lineage tracing, spatial transcriptomics, and organoid explant systems that model peripheral monocyte infiltration, we show that these programs are driven by microenvironmental cues and therapies rather than myeloid cell type, origin, or mutation status. The C1Q Immunosuppressive program is driven by routinely administered dexamethasone. The Scavenger Immunosuppressive program includes ligands with established roles in T-cell suppression, is induced in hypoxic regions, and is associated with immunotherapy resistance. Both immunosuppressive programs are less prevalent in lower-grade gliomas, which are instead enriched for the CXCR4 Inflammatory program. Our study provides a framework to understand immunomodulatory myeloid cells in glioma, and a foundation to develop more effective immunotherapies.
Conflict of interest statement
Competing interests T.E.M. discloses financial interest in Reify Health, Care Access Research, and Telomere Diagnostics. C.P.C. reports compensation for consulting from Axoft inc. L.N.G.C. reports consulting fees from Elsevier, Oakstone Publishing and BMJ Best Practice, and research funding from Merck & Co (to DFCI). J.L.G. is consultant/serves on the Scientific Advisory Board of Array BioPharma, AstraZeneca, BD Biosciences, Carisma, Codagenix, Duke Street Bio, GlaxoSmithKline, Kowa, Kymera, OncoOne, and Verseau Therapeutics, and receives research support from Array BioPharma/Pfizer, Eli Lilly, GlaxoSmithKline, and Merck. M.L.S. is an equity holder, scientific co-founder and advisory board member of Immunitas Therapeutics. A.K.S. reports compensation for consulting and/or scientific advisory board membership from Honeycomb Biotechnologies, Cellarity, Ochre Bio, Relation Therapeutics, FL86, IntrECate Biotherapeutics, Senda Biosciences and Dahlia Biosciences unrelated to this work. B.E.B. discloses financial interests in Fulcrum Therapeutics, HiFiBio, Arsenal Biosciences, Chroma Medicine, Cell Signaling Technologies, and Design Pharmaceuticals.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous