

This is a preprint.
A Latent Activated Olfactory Stem Cell State Revealed by Single-Cell Transcriptomic and Epigenomic Profiling
- PMID: 37961539
- PMCID: PMC10634988
- DOI: 10.1101/2023.10.26.564041
A Latent Activated Olfactory Stem Cell State Revealed by Single-Cell Transcriptomic and Epigenomic Profiling
Abstract
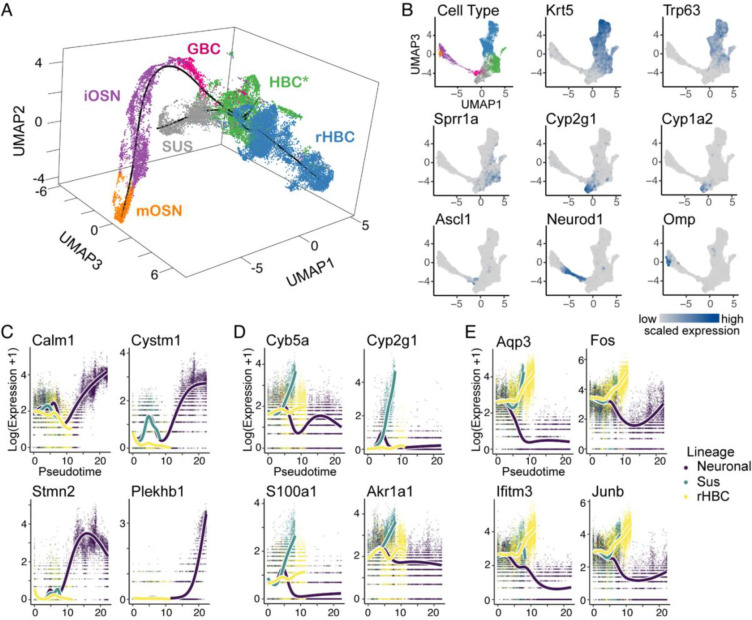
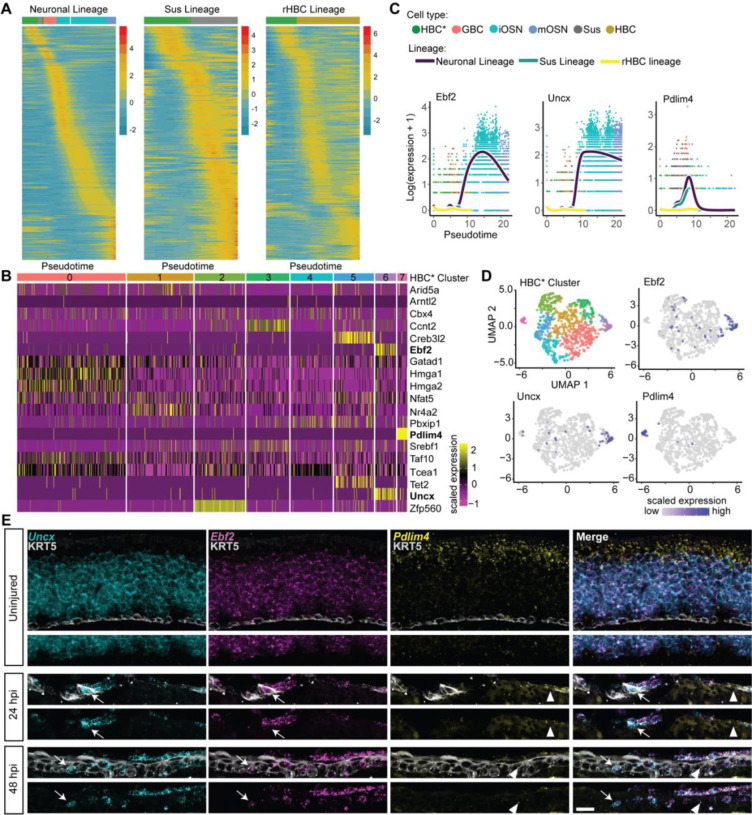
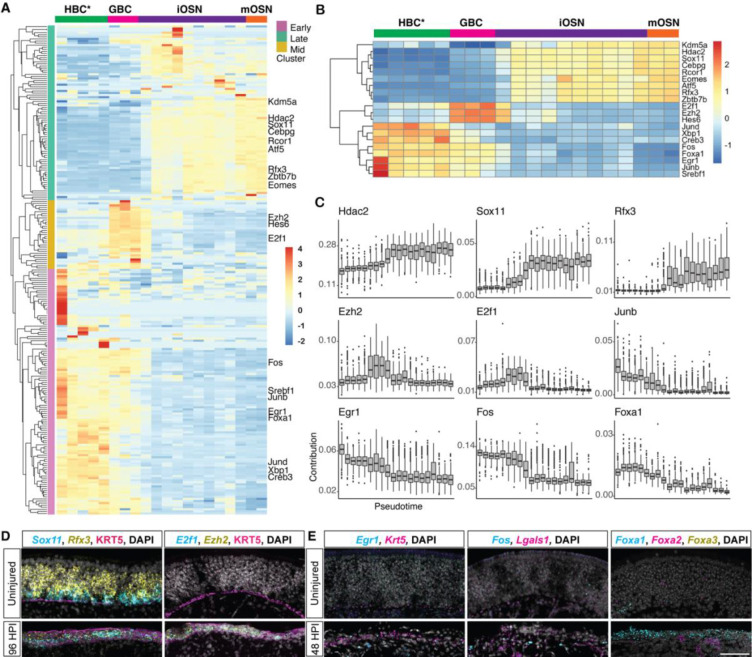
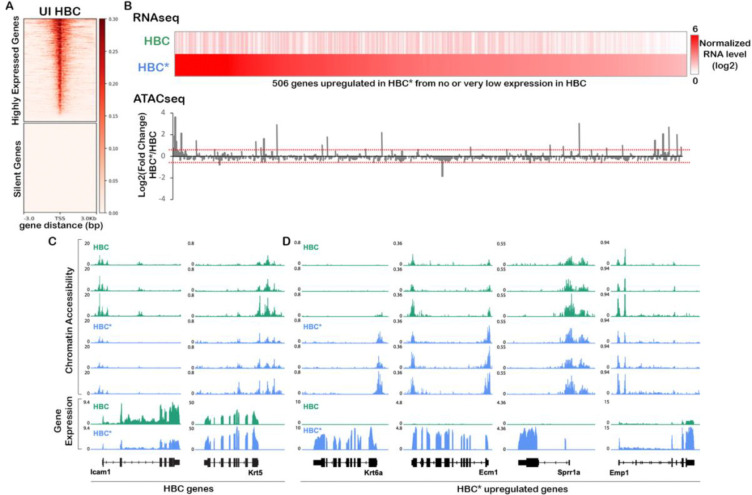
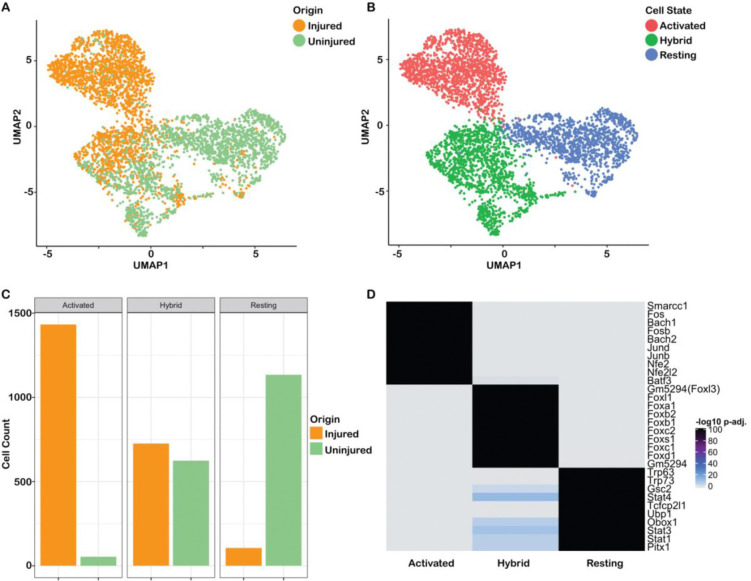
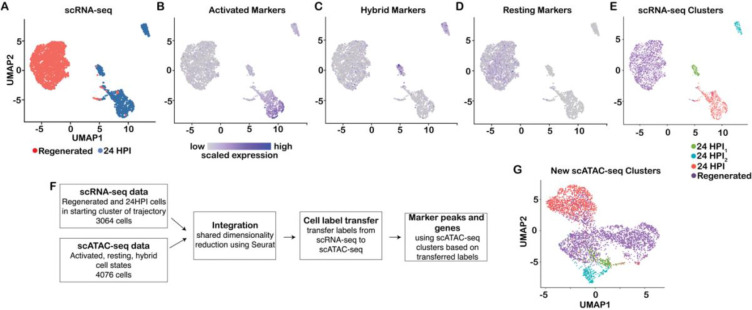
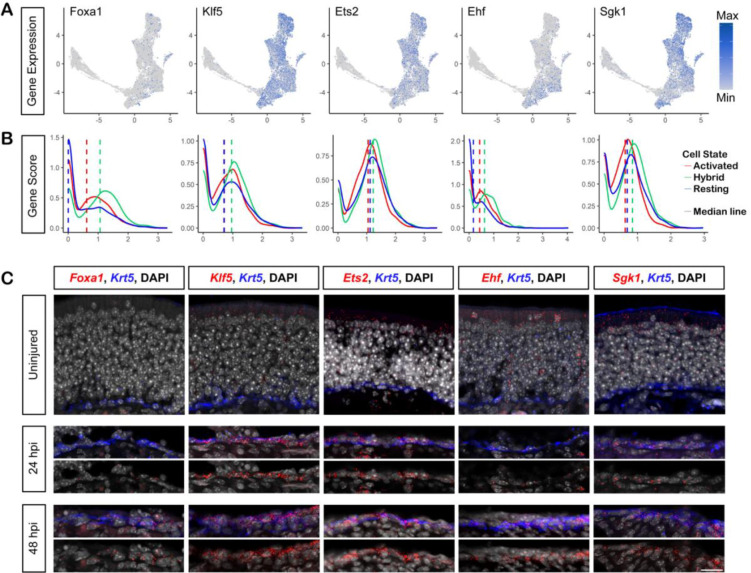
The olfactory epithelium is one of the few regions of the nervous system that sustains neurogenesis throughout life. Its experimental accessibility makes it especially tractable for studying molecular mechanisms that drive neural regeneration in response to injury. In this study, we used single-cell sequencing to identify the transcriptional cascades and epigenetic processes involved in determining olfactory epithelial stem cell fate during injury-induced regeneration. By combining gene expression and accessible chromatin profiles of individual lineage-traced olfactory stem cells, we identified transcriptional heterogeneity among activated stem cells at a stage when cell fates are being specified. We further identified a subset of resting cells that appears poised for activation, characterized by accessible chromatin around wound response and lineage-specific genes prior to their later expression in response to injury. Together these results provide evidence for a latent activated stem cell state, in which a subset of quiescent olfactory epithelial stem cells are epigenetically primed to support injury-induced regeneration.
Figures
Similar articles
-
Injury Activates Transient Olfactory Stem Cell States with Diverse Lineage Capacities.Cell Stem Cell. 2017 Dec 7;21(6):775-790.e9. doi: 10.1016/j.stem.2017.10.014. Epub 2017 Nov 22. Cell Stem Cell. 2017. PMID: 29174333 Free PMC article.
-
Chemokine signaling is required for homeostatic and injury-induced neurogenesis in the olfactory epithelium.Stem Cells. 2021 May;39(5):617-635. doi: 10.1002/stem.3338. Epub 2021 Jan 25. Stem Cells. 2021. PMID: 33470495
-
Canonical Notch Signaling Directs the Fate of Differentiating Neurocompetent Progenitors in the Mammalian Olfactory Epithelium.J Neurosci. 2018 May 23;38(21):5022-5037. doi: 10.1523/JNEUROSCI.0484-17.2018. Epub 2018 May 8. J Neurosci. 2018. PMID: 29739871 Free PMC article.
-
Identification and molecular regulation of neural stem cells in the olfactory epithelium.Exp Cell Res. 2005 Jun 10;306(2):309-16. doi: 10.1016/j.yexcr.2005.03.027. Epub 2005 Apr 21. Exp Cell Res. 2005. PMID: 15925585 Review.
-
Transcriptional and Epigenetic Control of Mammalian Olfactory Epithelium Development.Mol Neurobiol. 2018 Nov;55(11):8306-8327. doi: 10.1007/s12035-018-0987-y. Epub 2018 Mar 12. Mol Neurobiol. 2018. PMID: 29532253 Review.
References
-
- Azuara Véronique, Perry Pascale, Sauer Stephan, Spivakov Mikhail, Jørgensen Helle F., John Rosalind M., Gouti Mina, et al. 2006. “Chromatin Signatures of Pluripotent Cell Lines.” Nature Cell Biology 8 (5): 532–38. - PubMed
-
- Benjamini Yoav, and Hochberg Yosef. 1995. “Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing.” Journal of the Royal Statistical Society. Series B, Statistical Methodology 57 (1): 289–300.
-
- Bernstein Bradley E., Mikkelsen Tarjei S., Xie Xiaohui, Kamal Michael, Huebert Dana J., Cuff James, Fry Ben, et al. 2006. “A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells.” Cell 125 (2): 315–26. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases