

This is a preprint.
OASIS: An interpretable, finite-sample valid alternative to Pearson's for scientific discovery
- PMID: 37961606
- PMCID: PMC10634974
- DOI: 10.1101/2023.03.16.533008
OASIS: An interpretable, finite-sample valid alternative to Pearson's for scientific discovery
Update in
-
OASIS: An interpretable, finite-sample valid alternative to Pearson's X2 for scientific discovery.Proc Natl Acad Sci U S A. 2024 Apr 9;121(15):e2304671121. doi: 10.1073/pnas.2304671121. Epub 2024 Apr 2. Proc Natl Acad Sci U S A. 2024. PMID: 38564640 Free PMC article.
Abstract
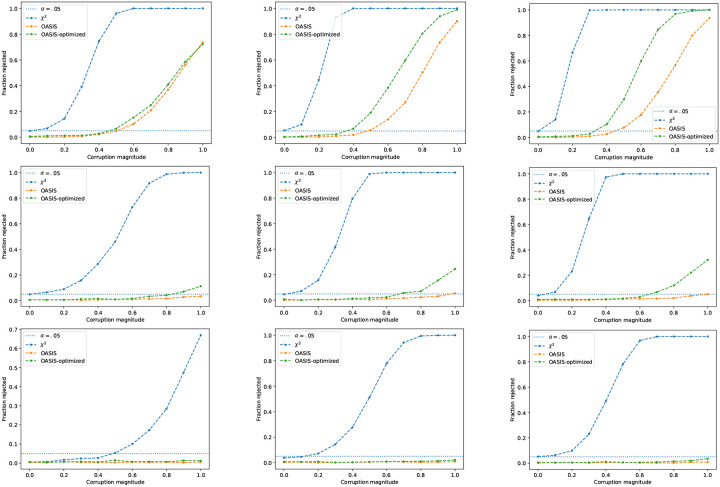
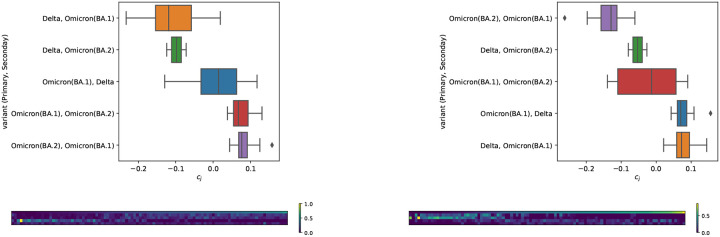
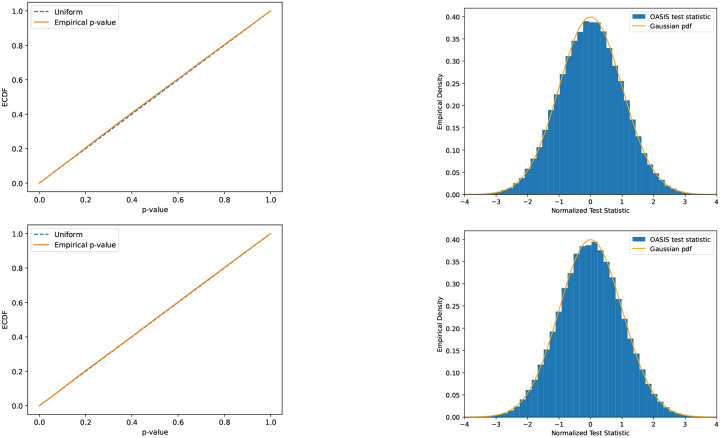
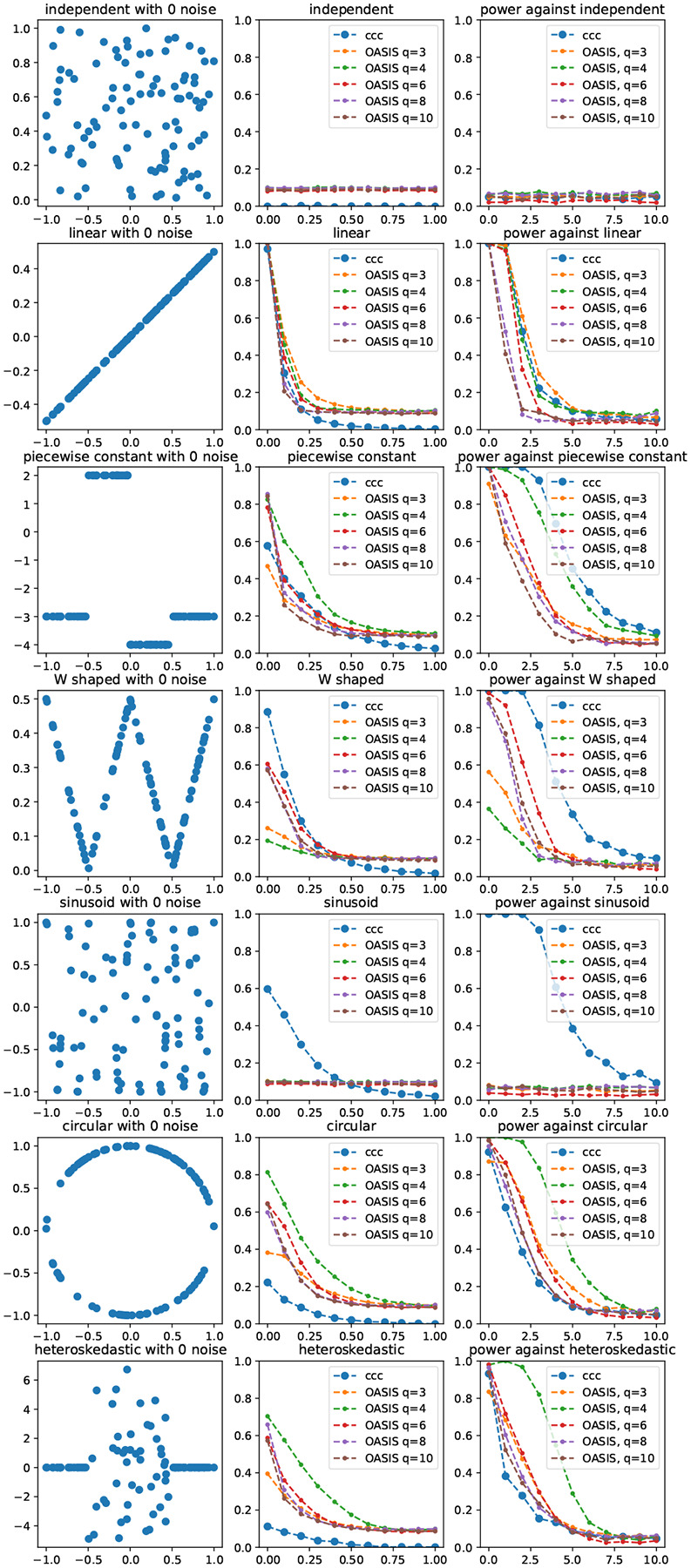
Contingency tables, data represented as counts matrices, are ubiquitous across quantitative research and data-science applications. Existing statistical tests are insufficient however, as none are simultaneously computationally efficient and statistically valid for a finite number of observations. In this work, motivated by a recent application in reference-free genomic inference (1), we develop OASIS (Optimized Adaptive Statistic for Inferring Structure), a family of statistical tests for contingency tables. OASIS constructs a test-statistic which is linear in the normalized data matrix, providing closed form p-value bounds through classical concentration inequalities. In the process, OASIS provides a decomposition of the table, lending interpretability to its rejection of the null. We derive the asymptotic distribution of the OASIS test statistic, showing that these finite-sample bounds correctly characterize the test statistic's p-value up to a variance term. Experiments on genomic sequencing data highlight the power and interpretability of OASIS. The same method based on OASIS significance calls detects SARS-CoV-2 and Mycobacterium Tuberculosis strains de novo, which cannot be achieved with current approaches. We demonstrate in simulations that OASIS is robust to overdispersion, a common feature in genomic data like single cell RNA-sequencing, where under accepted noise models OASIS still provides good control of the false discovery rate, while Pearson's test consistently rejects the null. Additionally, we show on synthetic data that OASIS is more powerful than Pearson's test in certain regimes, including for some important two group alternatives, which we corroborate with approximate power calculations.
Keywords: Alignment-free inference; Computational genomics; Contingency table; Finite-sample p-value bound.
Figures

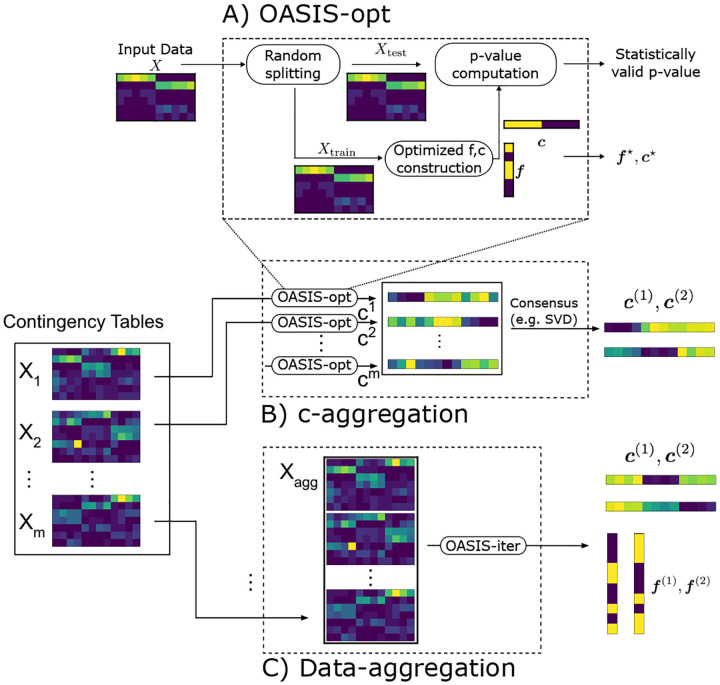
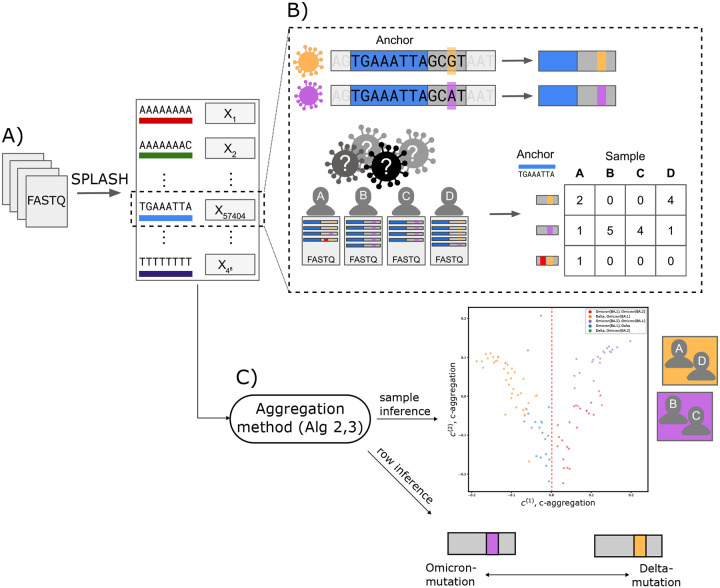



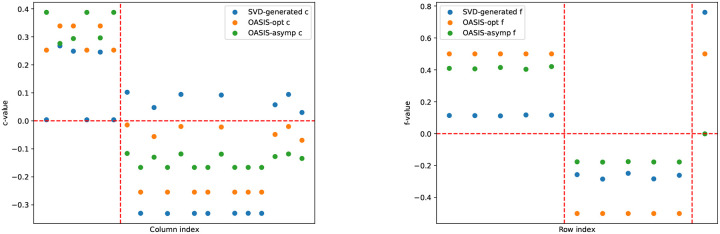




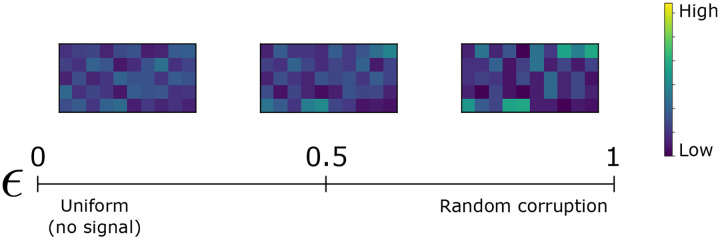













References
-
- Chaung K, Baharav T, Zheludev I, Salzman J, A statistical, reference-free algorithm subsumes myriad problems in genome science and enables novel discovery. bioRxiv (2022).
-
- Chen Y, Diaconis P, Holmes SP, Liu JS, Sequential monte carlo methods for statistical analysis of tables. J. Am. Stat. Assoc. 100, 109–120 (2005).
-
- Pearson K, On the criterion that a given system of deviations from the probable in the case of a correlated system of variables is such that it can be reasonably supposed to have arisen from random sampling. The London, Edinburgh, Dublin Philos. Mag. J. Sci. 50, 157–175 (1900).
-
- Agresti A, Categorical data analysis. (John Wiley & Sons; ) Vol. 792, (2012).
-
- Diaconis P, Sturmfels B, Algebraic algorithms for sampling from conditional distributions. The Annals statistics 26, 363–397 (1998).
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous