

Novel Pan-ERR Agonists Ameliorate Heart Failure Through Enhancing Cardiac Fatty Acid Metabolism and Mitochondrial Function
- PMID: 37961903
- PMCID: PMC10842599
- DOI: 10.1161/CIRCULATIONAHA.123.066542
Novel Pan-ERR Agonists Ameliorate Heart Failure Through Enhancing Cardiac Fatty Acid Metabolism and Mitochondrial Function
Abstract
Background: Cardiac metabolic dysfunction is a hallmark of heart failure (HF). Estrogen-related receptors ERRα and ERRγ are essential regulators of cardiac metabolism. Therefore, activation of ERR could be a potential therapeutic intervention for HF. However, in vivo studies demonstrating the potential usefulness of ERR agonist for HF treatment are lacking, because compounds with pharmacokinetics appropriate for in vivo use have not been available.
Methods: Using a structure-based design approach, we designed and synthesized 2 structurally distinct pan-ERR agonists, SLU-PP-332 and SLU-PP-915. We investigated the effect of ERR agonist on cardiac function in a pressure overload-induced HF model in vivo. We conducted comprehensive functional, multi-omics (RNA sequencing and metabolomics studies), and genetic dependency studies both in vivo and in vitro to dissect the molecular mechanism, ERR isoform dependency, and target specificity.
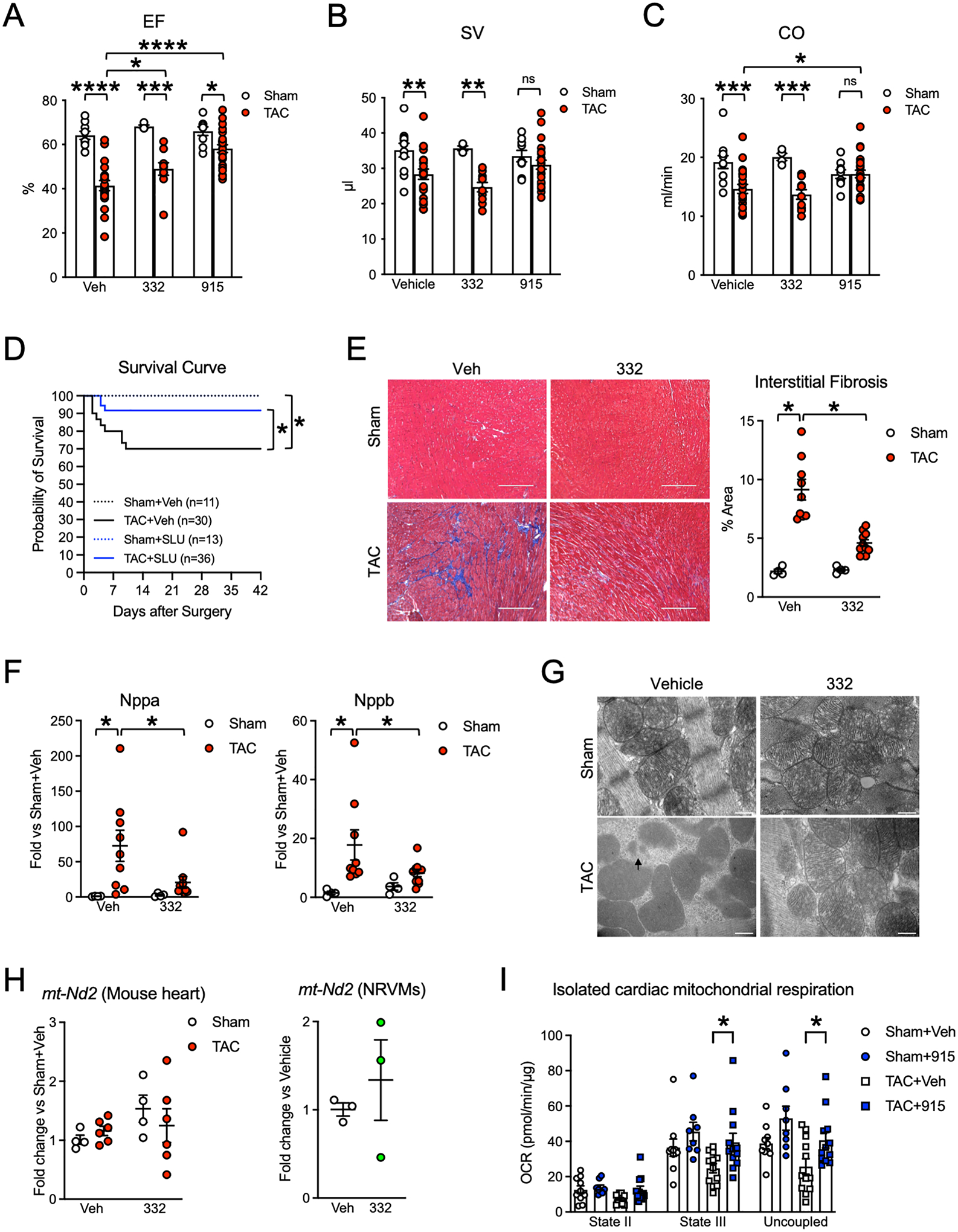
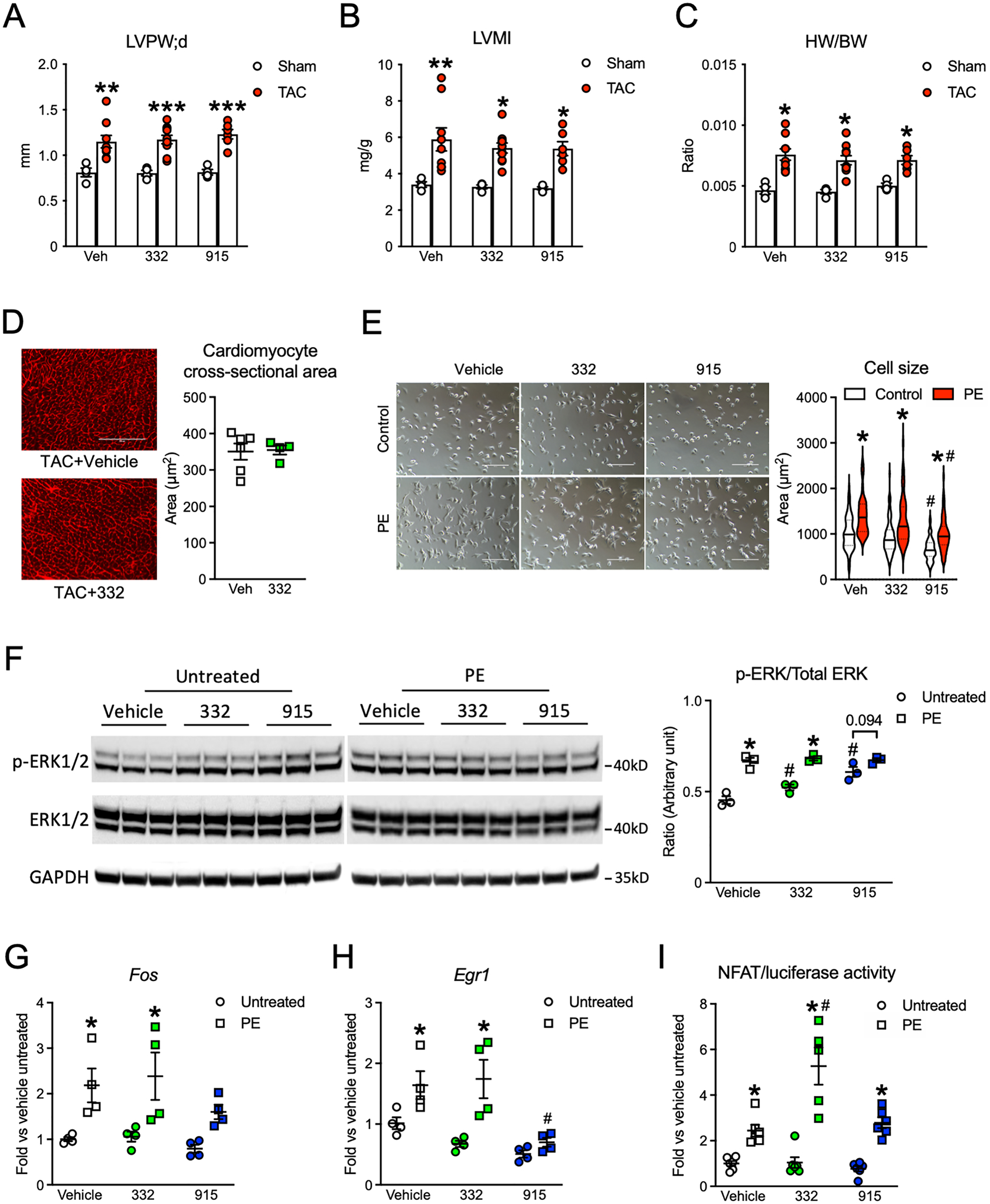
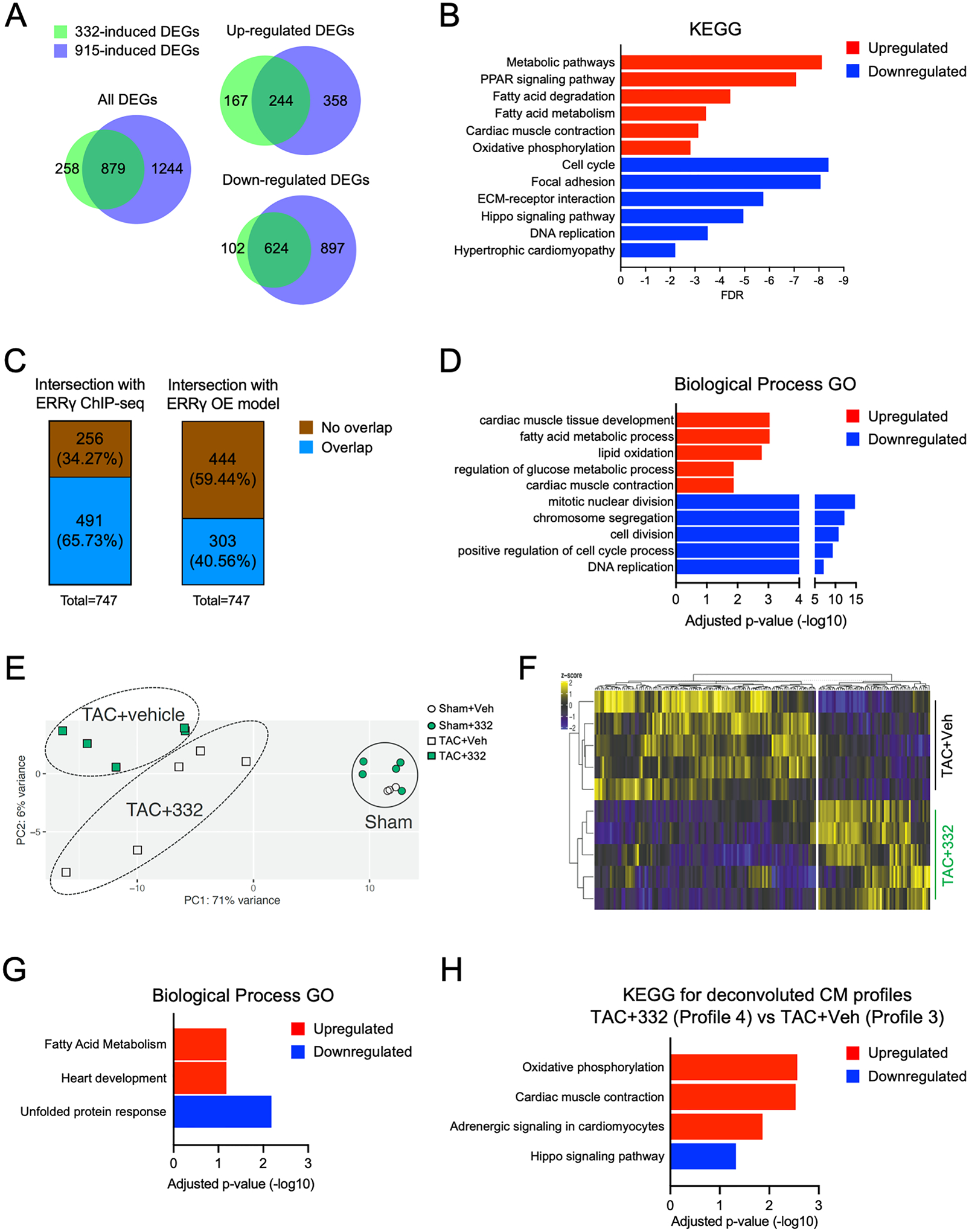
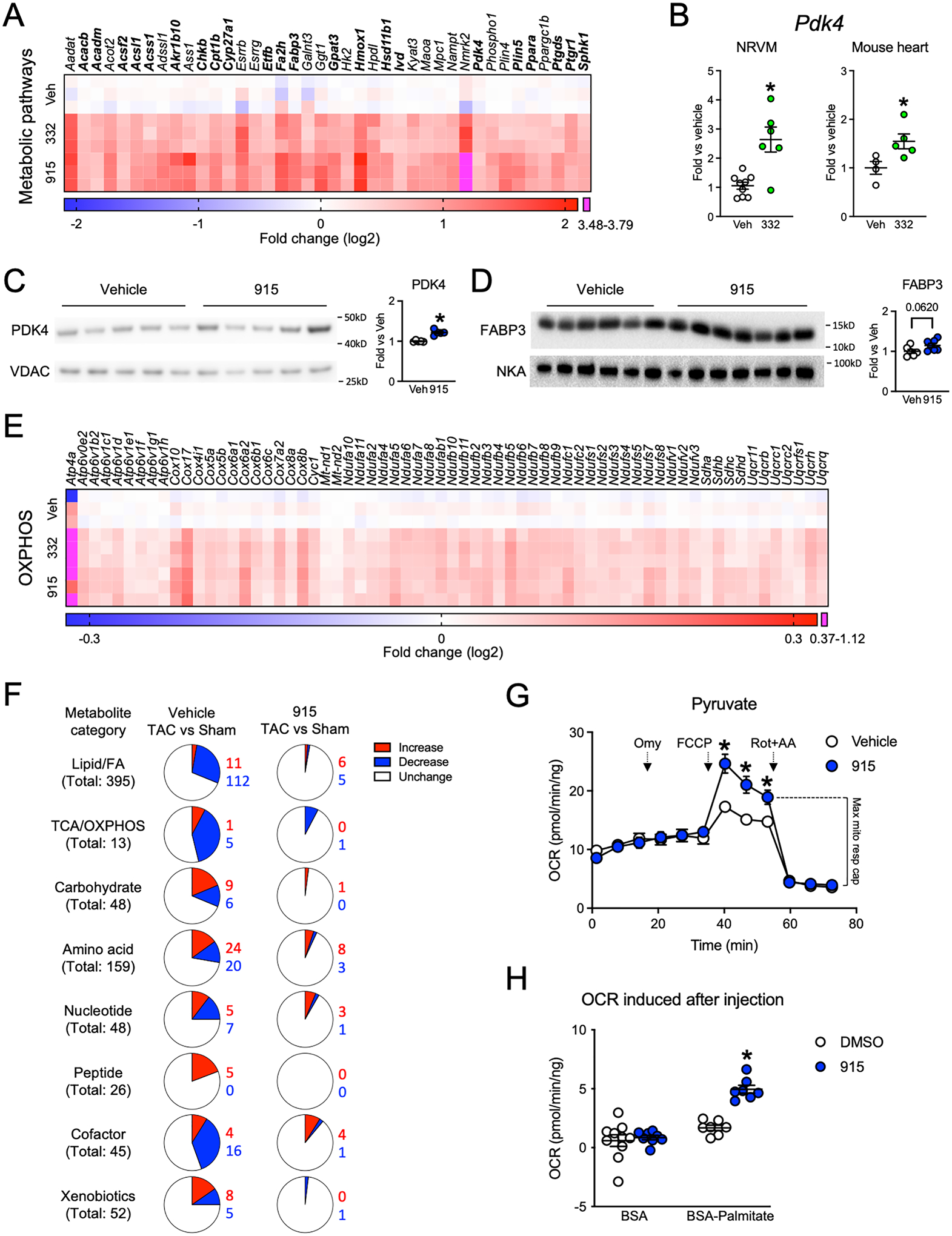
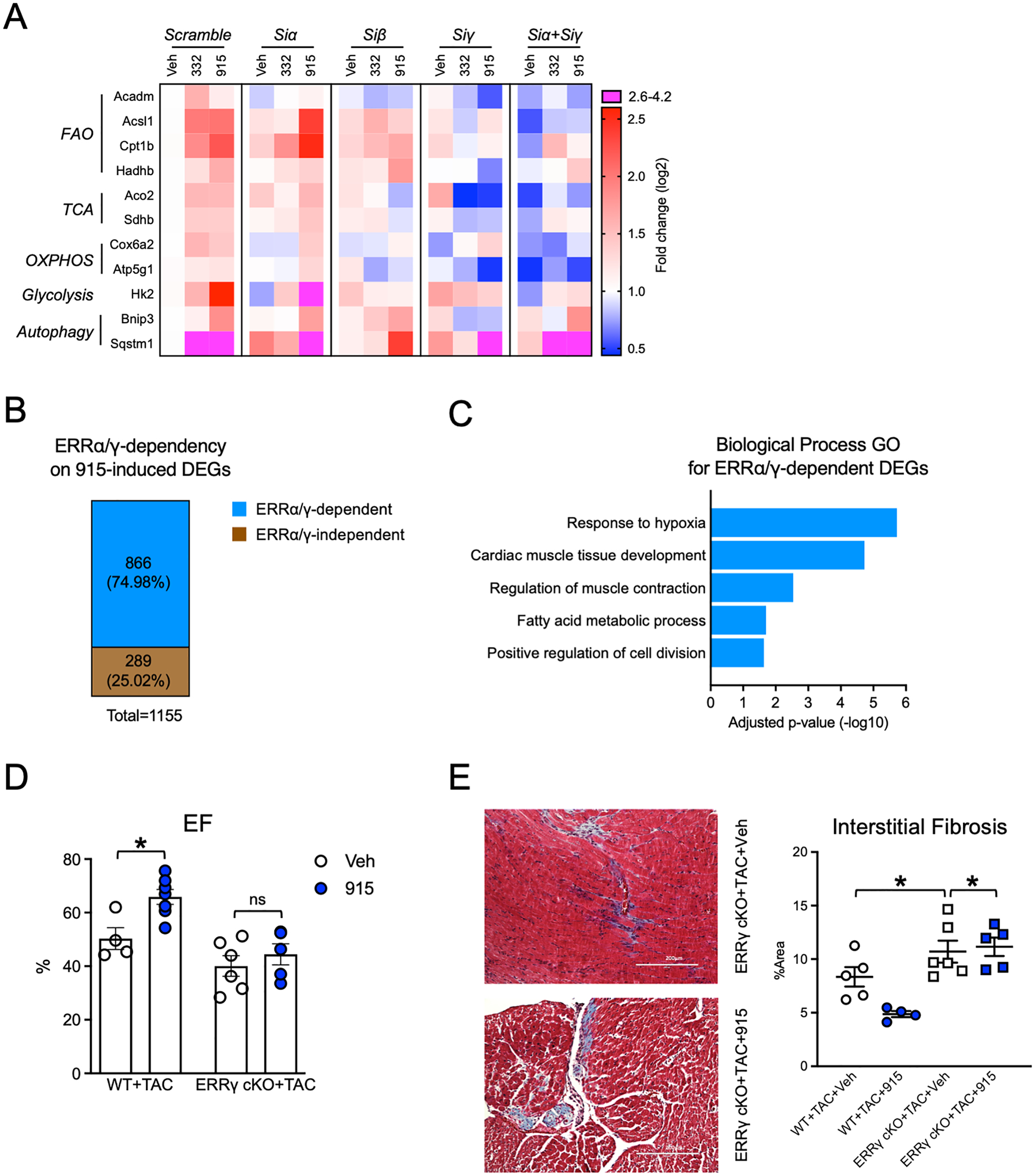
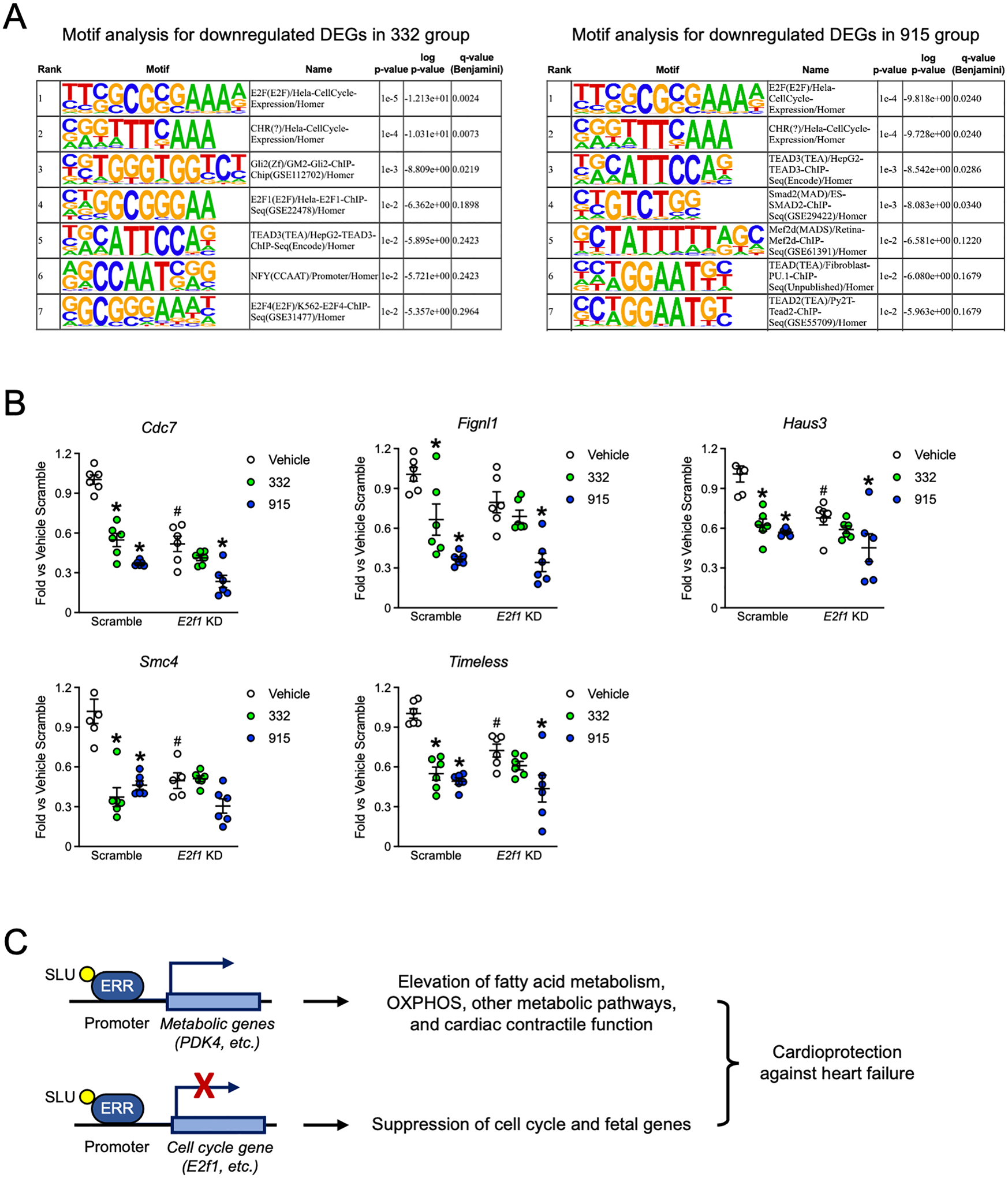
Results: Both SLU-PP-332 and SLU-PP-915 significantly improved ejection fraction, ameliorated fibrosis, and increased survival associated with pressure overload-induced HF without affecting cardiac hypertrophy. A broad spectrum of metabolic genes was transcriptionally activated by ERR agonists, particularly genes involved in fatty acid metabolism and mitochondrial function. Metabolomics analysis showed substantial normalization of metabolic profiles in fatty acid/lipid and tricarboxylic acid/oxidative phosphorylation metabolites in the mouse heart with 6-week pressure overload. ERR agonists increase mitochondria oxidative capacity and fatty acid use in vitro and in vivo. Using both in vitro and in vivo genetic dependency experiments, we show that ERRγ is the main mediator of ERR agonism-induced transcriptional regulation and cardioprotection and definitively demonstrated target specificity. ERR agonism also led to downregulation of cell cycle and development pathways, which was partially mediated by E2F1 in cardiomyocytes.
Conclusions: ERR agonists maintain oxidative metabolism, which confers cardiac protection against pressure overload-induced HF in vivo. Our results provide direct pharmacologic evidence supporting the further development of ERR agonists as novel HF therapeutics.
Keywords: cell cycle; heart failure; metabolism.
Conflict of interest statement
Figures
References
-
- Murphy SP, Ibrahim NE and Januzzi JL, Jr. Heart Failure With Reduced Ejection Fraction: A Review. JAMA. 2020;324:488–504. - PubMed
-
- Heidenreich PA, Albert NM, Allen LA, Bluemke DA, Butler J, Fonarow GC, Ikonomidis JS, Khavjou O, Konstam MA, Maddox TM, Nichol G, Pham M, Pina IL, Trogdon JG, American Heart Association Advocacy Coordinating C, Council on Arteriosclerosis T, Vascular B, Council on Cardiovascular R, Intervention, Council on Clinical C, Council on E, Prevention and Stroke C. Forecasting the impact of heart failure in the United States: a policy statement from the American Heart Association. Circ Heart Fail. 2013;6:606–19. - PMC - PubMed
-
- Mazurek JA and Jessup M. Understanding Heart Failure. Heart Fail Clin. 2017;13:1–19. - PubMed
-
- Stanley WC, Recchia FA and Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–129. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
