

OMA orthology in 2024: improved prokaryote coverage, ancestral and extant GO enrichment, a revamped synteny viewer and more in the OMA Ecosystem
- PMID: 37962356
- PMCID: PMC10767875
- DOI: 10.1093/nar/gkad1020
OMA orthology in 2024: improved prokaryote coverage, ancestral and extant GO enrichment, a revamped synteny viewer and more in the OMA Ecosystem
Abstract
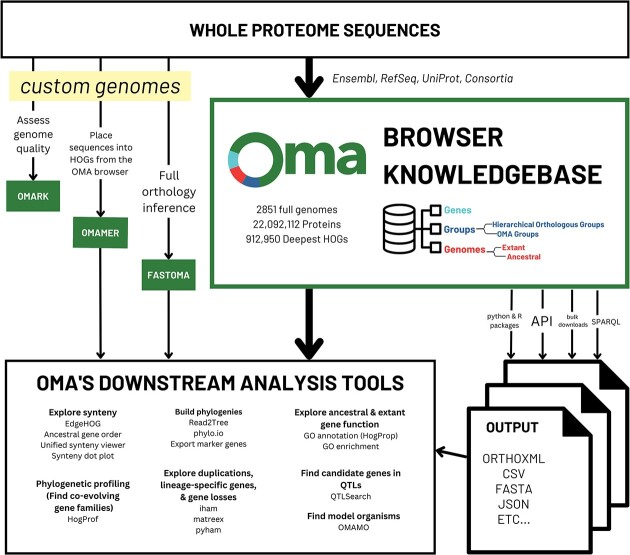
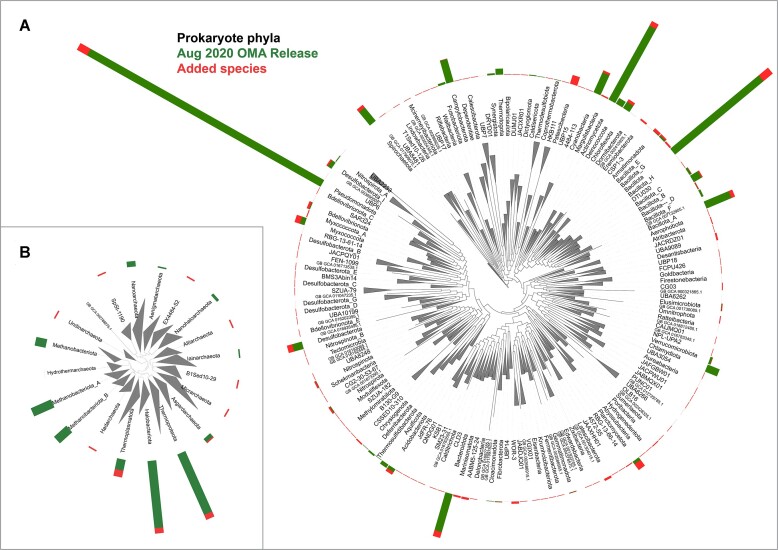
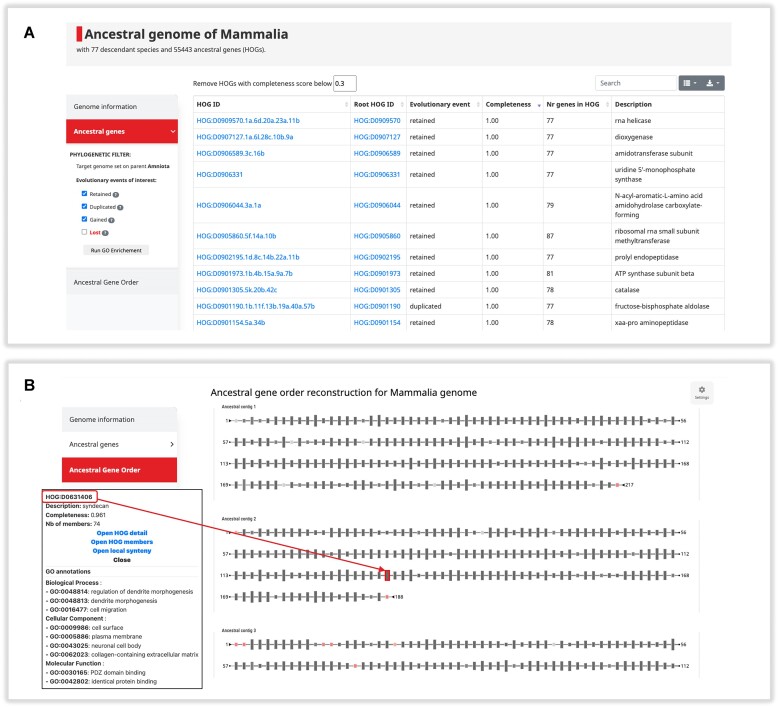
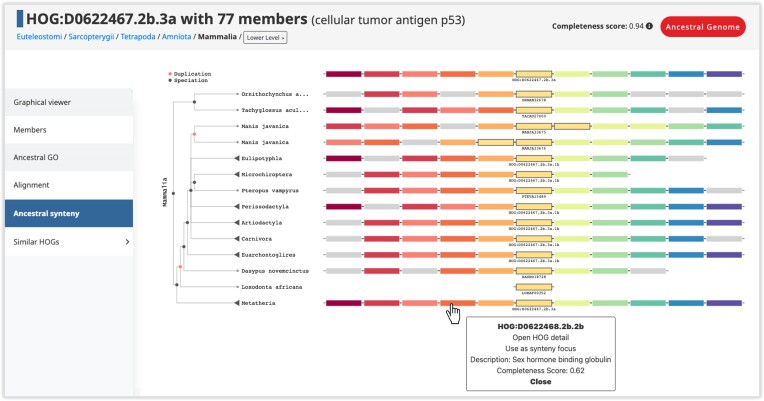
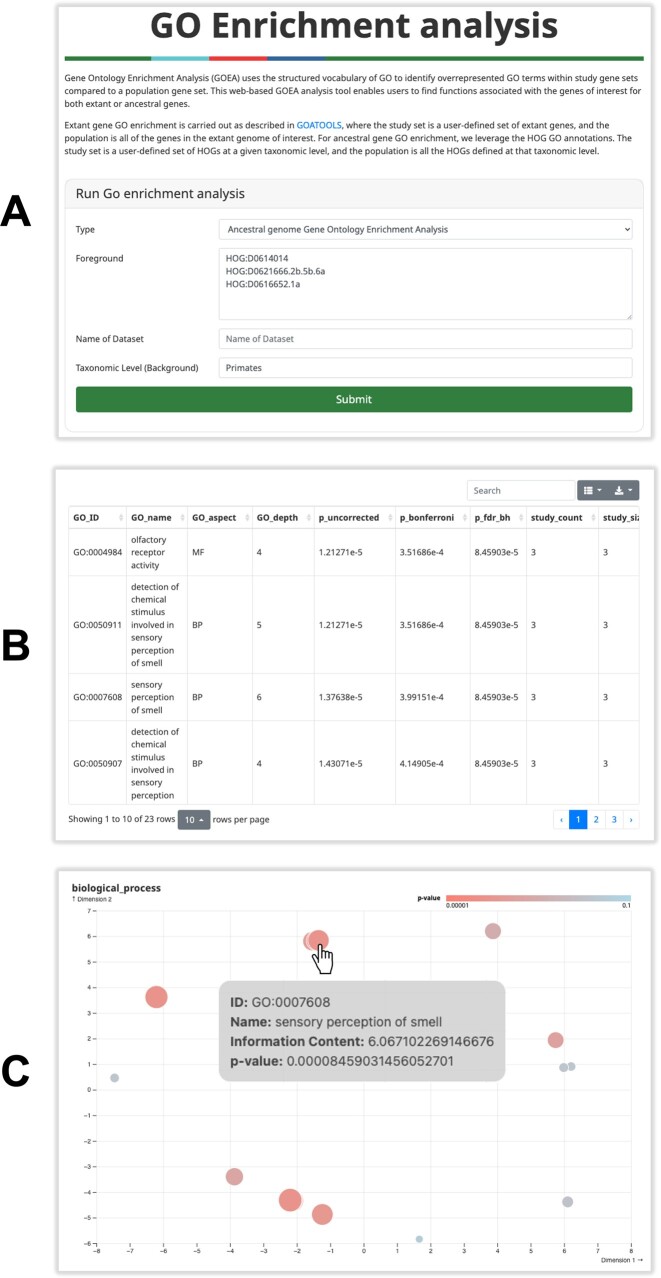
In this update paper, we present the latest developments in the OMA browser knowledgebase, which aims to provide high-quality orthology inferences and facilitate the study of gene families, genomes and their evolution. First, we discuss the addition of new species in the database, particularly an expanded representation of prokaryotic species. The OMA browser now offers Ancestral Genome pages and an Ancestral Gene Order viewer, allowing users to explore the evolutionary history and gene content of ancestral genomes. We also introduce a revamped Local Synteny Viewer to compare genomic neighborhoods across both extant and ancestral genomes. Hierarchical Orthologous Groups (HOGs) are now annotated with Gene Ontology annotations, and users can easily perform extant or ancestral GO enrichments. Finally, we recap new tools in the OMA Ecosystem, including OMAmer for proteome mapping, OMArk for proteome quality assessment, OMAMO for model organism selection and Read2Tree for phylogenetic species tree construction from reads. These new features provide exciting opportunities for orthology analysis and comparative genomics. OMA is accessible at https://omabrowser.org.
© The Author(s) 2023. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Fitch W.M. Distinguishing homologous from analogous proteins. Syst. Zool. 1970; 19:99–113. - PubMed
-
- Dessimoz C., Cannarozzi G., Gil M., Margadant D., Roth A., Schneider A., Gonnet G. McLysaght A., Huson D.H.. OMA, A comprehensive, automated project for the identification of orthologs from complete genome data: introduction and first achievements. RECOMB 2005 Workshop on Comparative Genomics. 2005; Springer-Verlag; 61–72.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
