

Survival and acute exacerbation for patients with idiopathic pulmonary fibrosis (IPF) or non-IPF idiopathic interstitial pneumonias: 5-year follow-up analysis of a prospective multi-institutional patient registry
- PMID: 37963676
- PMCID: PMC10649622
- DOI: 10.1136/bmjresp-2023-001864
Survival and acute exacerbation for patients with idiopathic pulmonary fibrosis (IPF) or non-IPF idiopathic interstitial pneumonias: 5-year follow-up analysis of a prospective multi-institutional patient registry
Abstract
Objective: Few prospective cohort studies with relatively large numbers of patients with non-idiopathic pulmonary fibrosis (non-IPF) of idiopathic interstitial pneumonia (IIP) have been described. We aimed to assess disease progression and cause of death for patients with non-IPF IIPs or IPF under real-life conditions.
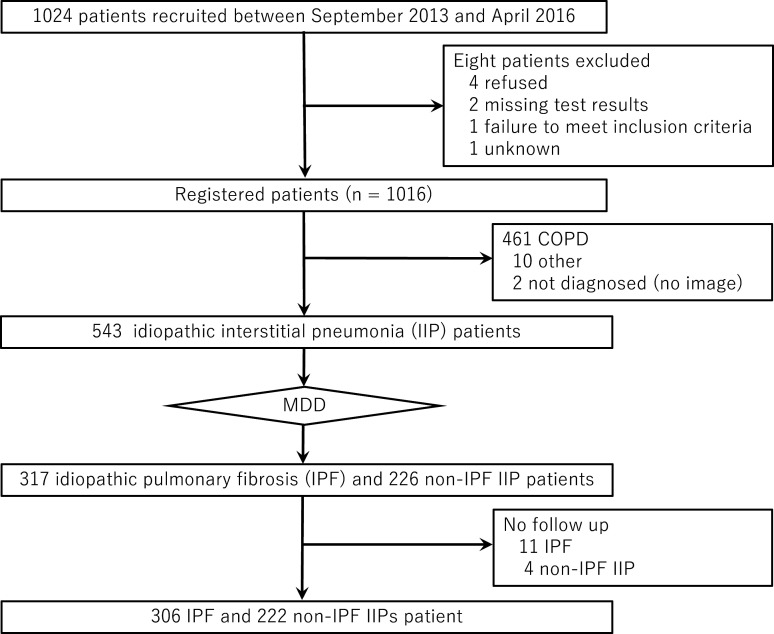
Methods: Data were analysed for a prospective multi-institutional cohort of 528 IIP patients enrolled in Japan between September 2013 and April 2016. Diagnosis of IPF versus non-IPF IIPs was based on central multidisciplinary discussion, and follow-up surveillance was performed for up to 5 years after patient registration. Survival and acute exacerbation (AE) were assessed.
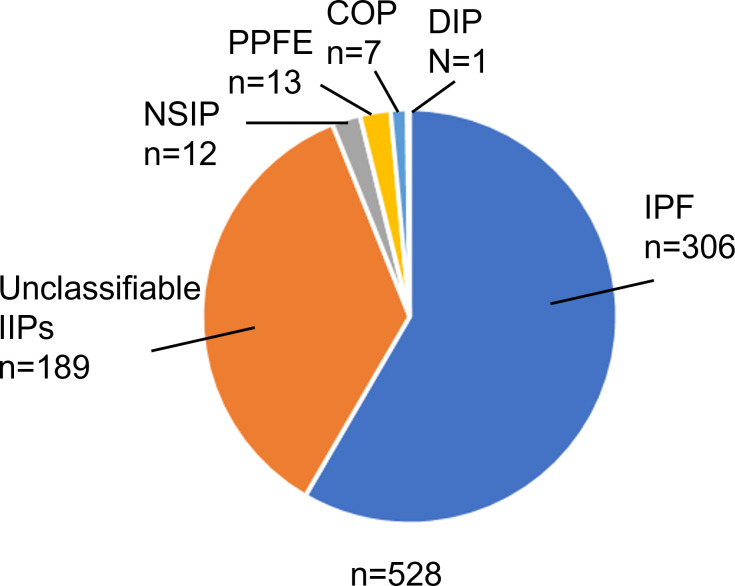
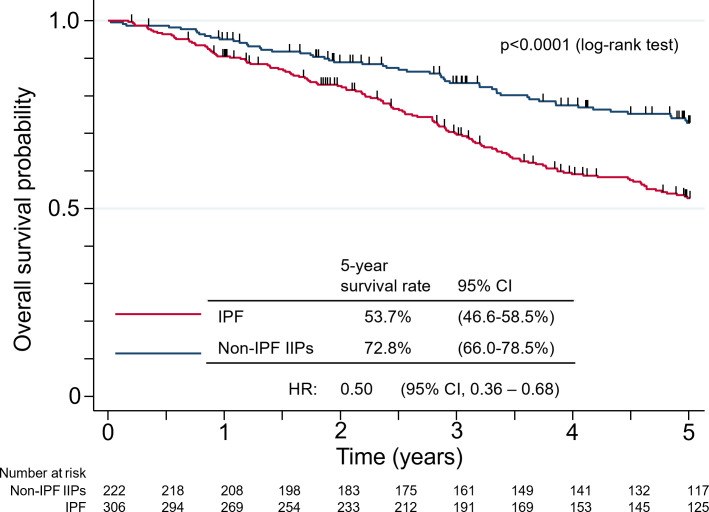
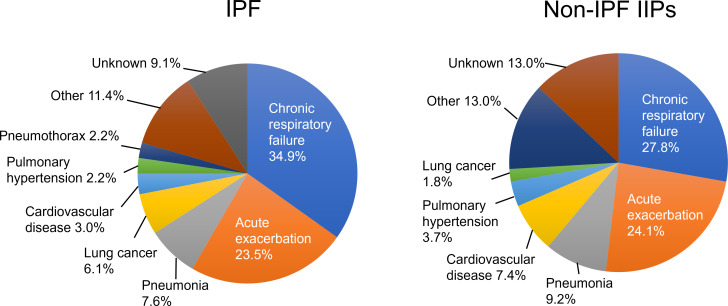
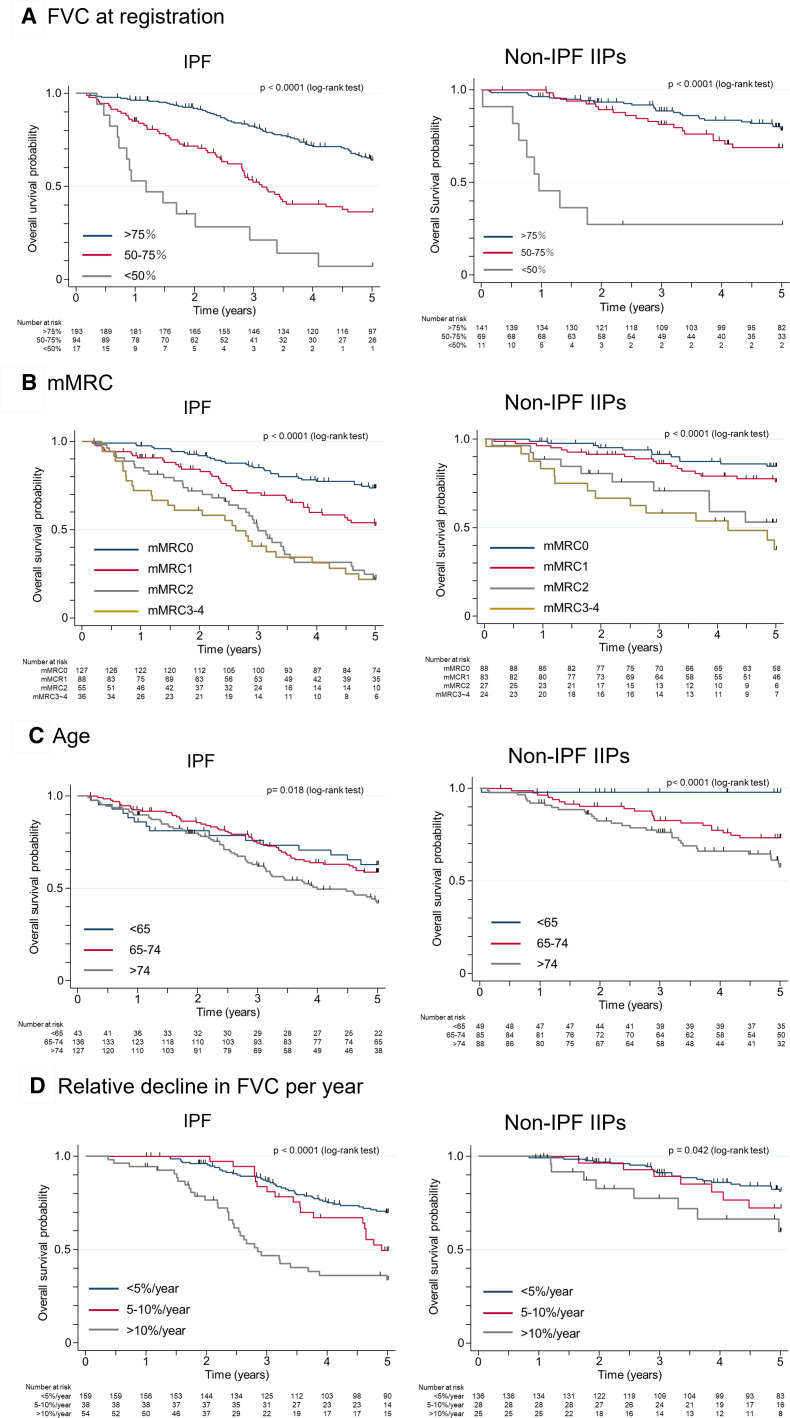
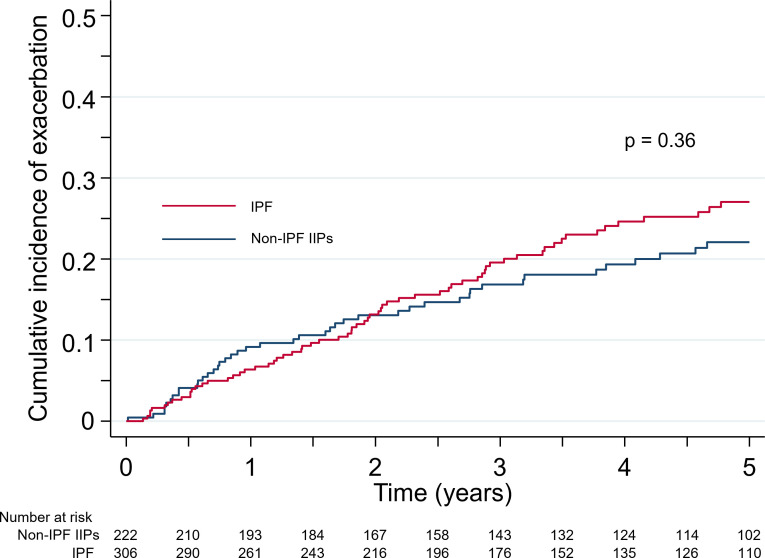
Results: IPF was the most common diagnosis (58.0%), followed by unclassifiable IIPs (35.8%) and others (6.2%). The 5-year survival rate for non-IPF IIP and IPF groups was 72.8% and 53.7%, respectively, with chronic respiratory failure being the primary cause of death in both groups. AE was the second most common cause of death for both non-IPF IIP (24.1%) and IPF (23.5%) patients. The cumulative incidence of AE did not differ significantly between the two groups (p=0.36), with a 1-year incidence rate of 7.4% and 9.0% in non-IPF IIP and IPF patients, respectively. We found that 30.2% and 39.4% of non-IPF IIP and IPF patients, respectively, who experienced AE died within 3 months after an AE event, whereas 55.8% and 66.7% of such patients, respectively, died within 5 years after registration.
Conclusion: Closer monitoring of disease progression and palliative care interventions after AE are important for non-IPF IIP patients as well as for IPF patients.
Keywords: Clinical Epidemiology; Interstitial Fibrosis.
© Author(s) (or their employer(s)) 2023. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: Yes.
Figures
Similar articles
-
Heterogeneity of incidence and outcome of acute exacerbation in idiopathic interstitial pneumonia.Respirology. 2016 Nov;21(8):1431-1437. doi: 10.1111/resp.12862. Epub 2016 Jul 26. Respirology. 2016. PMID: 27460223
-
Prognosis after acute exacerbation in patients with interstitial lung disease other than idiopathic pulmonary fibrosis.Clin Respir J. 2021 Mar;15(3):336-344. doi: 10.1111/crj.13304. Epub 2020 Dec 10. Clin Respir J. 2021. PMID: 33197284
-
Prognostic differences among patients with idiopathic interstitial pneumonias with acute exacerbation of varying pathogenesis: a retrospective study.Respir Res. 2019 Dec 18;20(1):287. doi: 10.1186/s12931-019-1247-z. Respir Res. 2019. PMID: 31852459 Free PMC article.
-
Incidence of acute exacerbation of interstitial pneumonia in operated lung cancer: institutional report and review.Ann Thorac Cardiovasc Surg. 2012;18(4):314-7. doi: 10.5761/atcs.oa.11.01839. Epub 2012 Mar 24. Ann Thorac Cardiovasc Surg. 2012. PMID: 22446955 Review.
-
Acute and subacute idiopathic interstitial pneumonias.Respirology. 2016 Jul;21(5):810-20. doi: 10.1111/resp.12786. Epub 2016 Apr 28. Respirology. 2016. PMID: 27123874 Review.
Cited by
-
Current state of signaling pathways associated with the pathogenesis of idiopathic pulmonary fibrosis.Respir Res. 2024 Jun 17;25(1):245. doi: 10.1186/s12931-024-02878-z. Respir Res. 2024. PMID: 38886743 Free PMC article. Review.
-
Update of prognosis and characteristics of chronic obstructive pulmonary disease in a real-world setting: a 5-year follow-up analysis of a multi-institutional registry.BMC Pulm Med. 2024 Nov 6;24(1):556. doi: 10.1186/s12890-024-03347-5. BMC Pulm Med. 2024. PMID: 39506773 Free PMC article.
-
Serum surfactant protein D as a significant biomarker for predicting occurrence, progression, acute exacerbation, and mortality in interstitial lung disease: a systematic review and meta-analysis.Front Immunol. 2025 Feb 14;16:1450798. doi: 10.3389/fimmu.2025.1450798. eCollection 2025. Front Immunol. 2025. PMID: 40028331 Free PMC article.
-
Treatment patterns and clinical profile in progressive pulmonary fibrosis: a Japanese cross-sectional survey.Front Med (Lausanne). 2025 Jan 15;11:1526531. doi: 10.3389/fmed.2024.1526531. eCollection 2024. Front Med (Lausanne). 2025. PMID: 39882519 Free PMC article.
-
Mortality and exacerbation risk according to GOLD and STAR severity stages of COPD: a 5-year multicenter prospective cohort study.Sci Rep. 2025 May 30;15(1):19097. doi: 10.1038/s41598-025-05033-w. Sci Rep. 2025. PMID: 40447720 Free PMC article.
References
-
- Travis WD, Costabel U, Hansell DM, et al. An official American thoracic society/European respiratory society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013;188:733–48. 10.1164/rccm.201308-1483ST - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous