

CHEX-seq detects single-cell genomic single-stranded DNA with catalytical potential
- PMID: 37963886
- PMCID: PMC10645931
- DOI: 10.1038/s41467-023-43158-6
CHEX-seq detects single-cell genomic single-stranded DNA with catalytical potential
Abstract
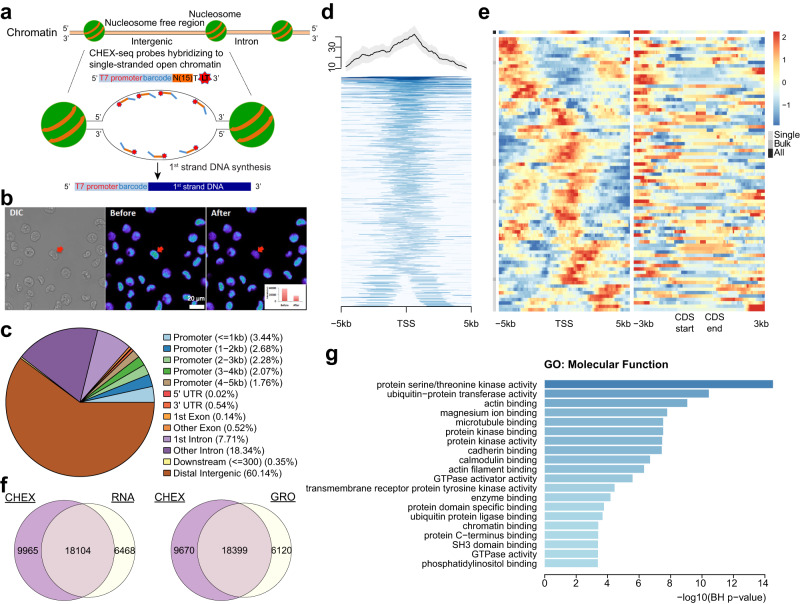
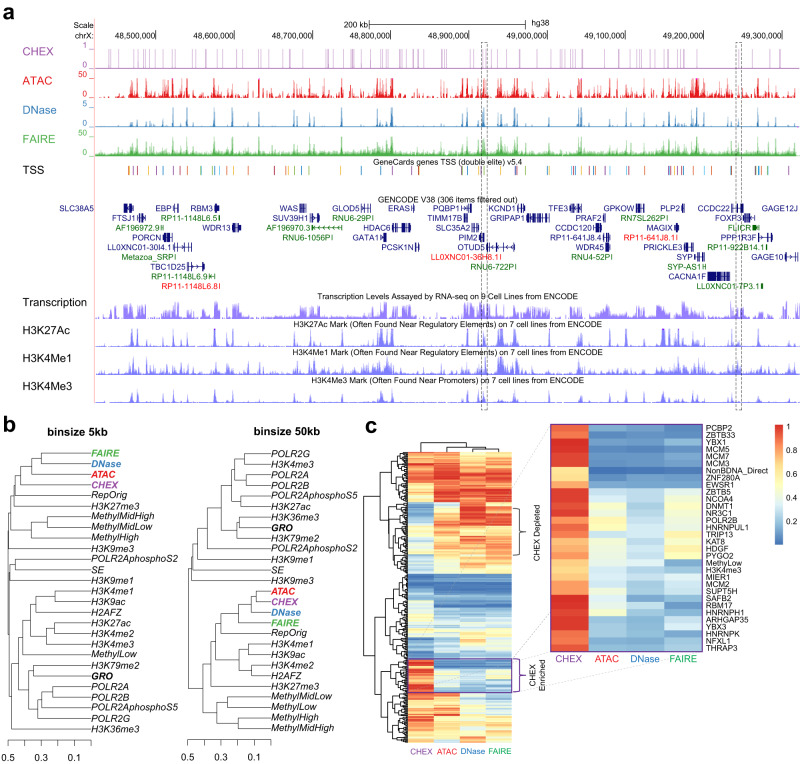
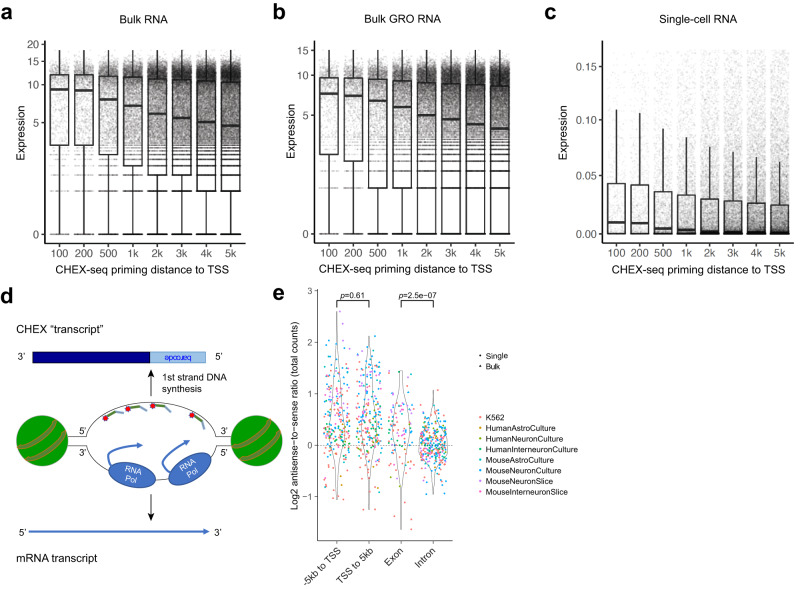
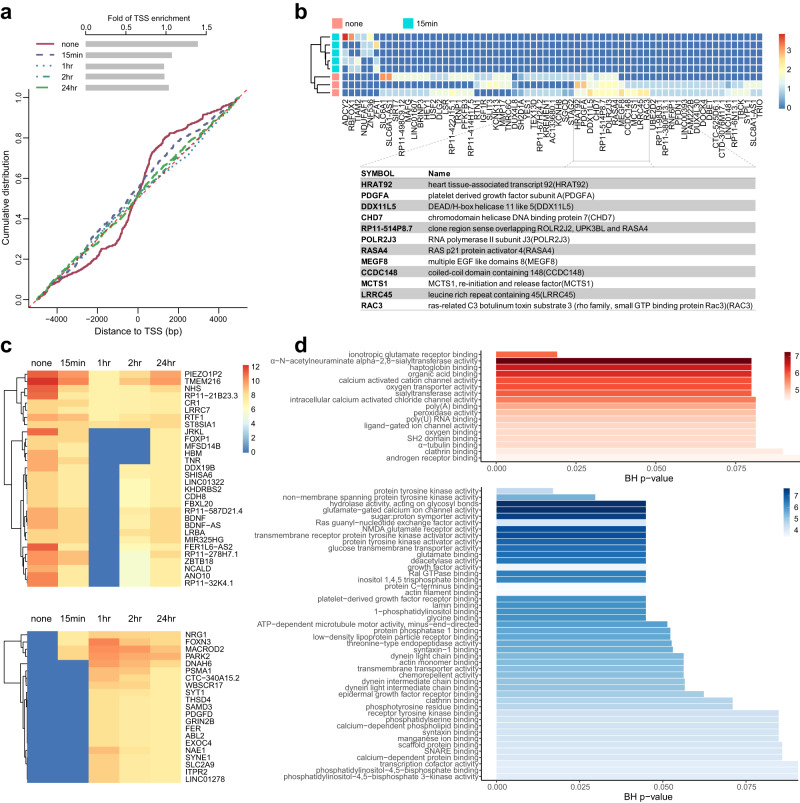
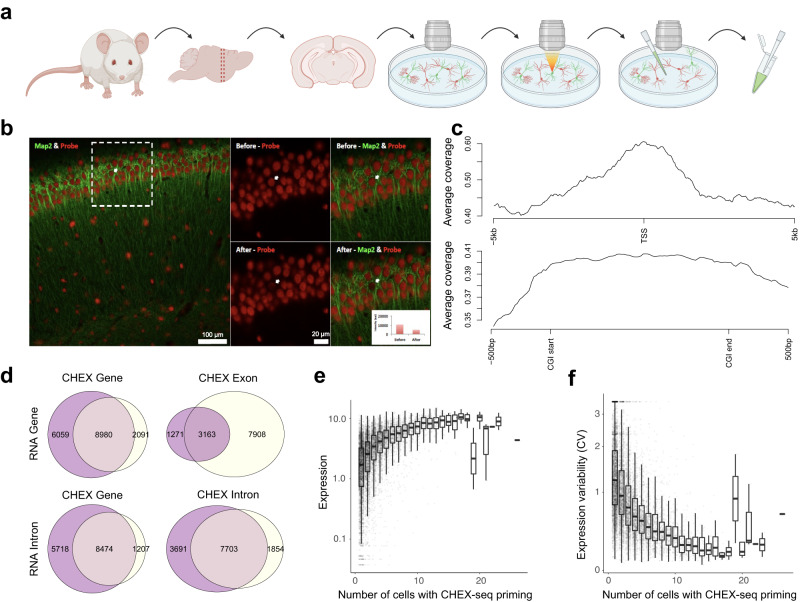
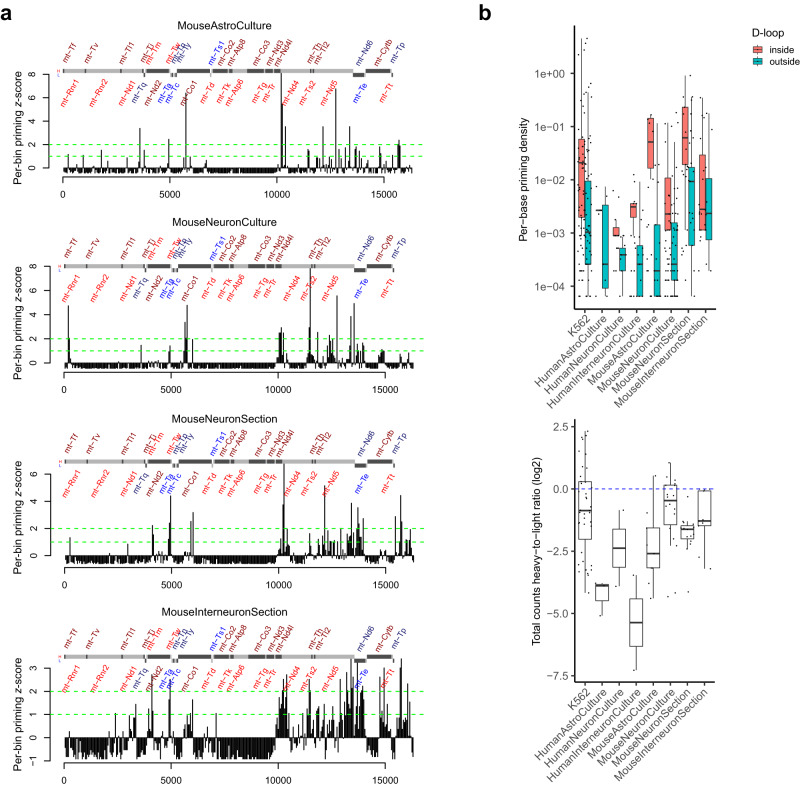
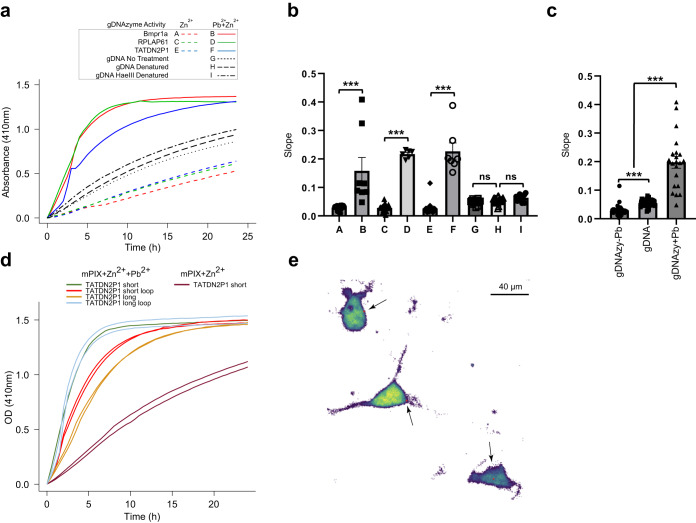
Genomic DNA (gDNA) undergoes structural interconversion between single- and double-stranded states during transcription, DNA repair and replication, which is critical for cellular homeostasis. We describe "CHEX-seq" which identifies the single-stranded DNA (ssDNA) in situ in individual cells. CHEX-seq uses 3'-terminal blocked, light-activatable probes to prime the copying of ssDNA into complementary DNA that is sequenced, thereby reporting the genome-wide single-stranded chromatin landscape. CHEX-seq is benchmarked in human K562 cells, and its utilities are demonstrated in cultures of mouse and human brain cells as well as immunostained spatially localized neurons in brain sections. The amount of ssDNA is dynamically regulated in response to perturbation. CHEX-seq also identifies single-stranded regions of mitochondrial DNA in single cells. Surprisingly, CHEX-seq identifies single-stranded loci in mouse and human gDNA that catalyze porphyrin metalation in vitro, suggesting a catalytic activity for genomic ssDNA. We posit that endogenous DNA enzymatic activity is a function of genomic ssDNA.
© 2023. The Author(s).
Conflict of interest statement
The Authors declare the following competing interests. J.E., J.Y.K., Y.L., S.F., J. Li, are co-inventors on a published patent application (20200216841) covering the CHEX-seq technology applied for by the University of Pennsylvania and Agilent Technologies. J.E., Y.L., J.Y.K., S.R.A., S.V. are co-inventors on a provisional patent application (63/511,984) covering gDNAzymes applied for by the University of Pennsylvania. The remaining authors declare no competing interests. This work was funded in part by NIH U01MH098953 (J.E., J.K.) and RM1HG010023 (J.K., J.E.), and by R01MH110185 (S.A).
Figures
Similar articles
-
Functions of single-strand DNA-binding proteins in DNA replication, recombination, and repair.Methods Mol Biol. 2012;922:1-21. doi: 10.1007/978-1-62703-032-8_1. Methods Mol Biol. 2012. PMID: 22976174
-
RADX interacts with single-stranded DNA to promote replication fork stability.EMBO Rep. 2017 Nov;18(11):1991-2003. doi: 10.15252/embr.201744877. Epub 2017 Oct 11. EMBO Rep. 2017. PMID: 29021206 Free PMC article.
-
Strand-specific ChIP-seq at DNA breaks distinguishes ssDNA versus dsDNA binding and refutes single-stranded nucleosomes.Mol Cell. 2021 Apr 15;81(8):1841-1853.e4. doi: 10.1016/j.molcel.2021.02.005. Epub 2021 Mar 1. Mol Cell. 2021. PMID: 33651987
-
Replication protein A: single-stranded DNA's first responder: dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair.Bioessays. 2014 Dec;36(12):1156-61. doi: 10.1002/bies.201400107. Epub 2014 Aug 29. Bioessays. 2014. PMID: 25171654 Free PMC article. Review.
-
Chaperoning RPA during DNA metabolism.Curr Genet. 2019 Aug;65(4):857-864. doi: 10.1007/s00294-019-00945-3. Epub 2019 Feb 22. Curr Genet. 2019. PMID: 30796471 Review.
Cited by
-
Base excision repair and double strand break repair cooperate to modulate the formation of unrepaired double strand breaks in mouse brain.Nat Commun. 2024 Sep 4;15(1):7726. doi: 10.1038/s41467-024-51906-5. Nat Commun. 2024. PMID: 39231940 Free PMC article.
-
Tandem ssDNA in neutrophil extracellular traps binds thrombin and regulates immunothrombosis.Proc Natl Acad Sci U S A. 2025 Jul 8;122(27):e2418191122. doi: 10.1073/pnas.2418191122. Epub 2025 Jul 3. Proc Natl Acad Sci U S A. 2025. PMID: 40608679
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
