

Long noncoding RNAs emerge from transposon-derived antisense sequences and may contribute to infection stage-specific transposon regulation in a fungal phytopathogen
- PMID: 37964319
- PMCID: PMC10648671
- DOI: 10.1186/s13100-023-00305-6
Long noncoding RNAs emerge from transposon-derived antisense sequences and may contribute to infection stage-specific transposon regulation in a fungal phytopathogen
Abstract
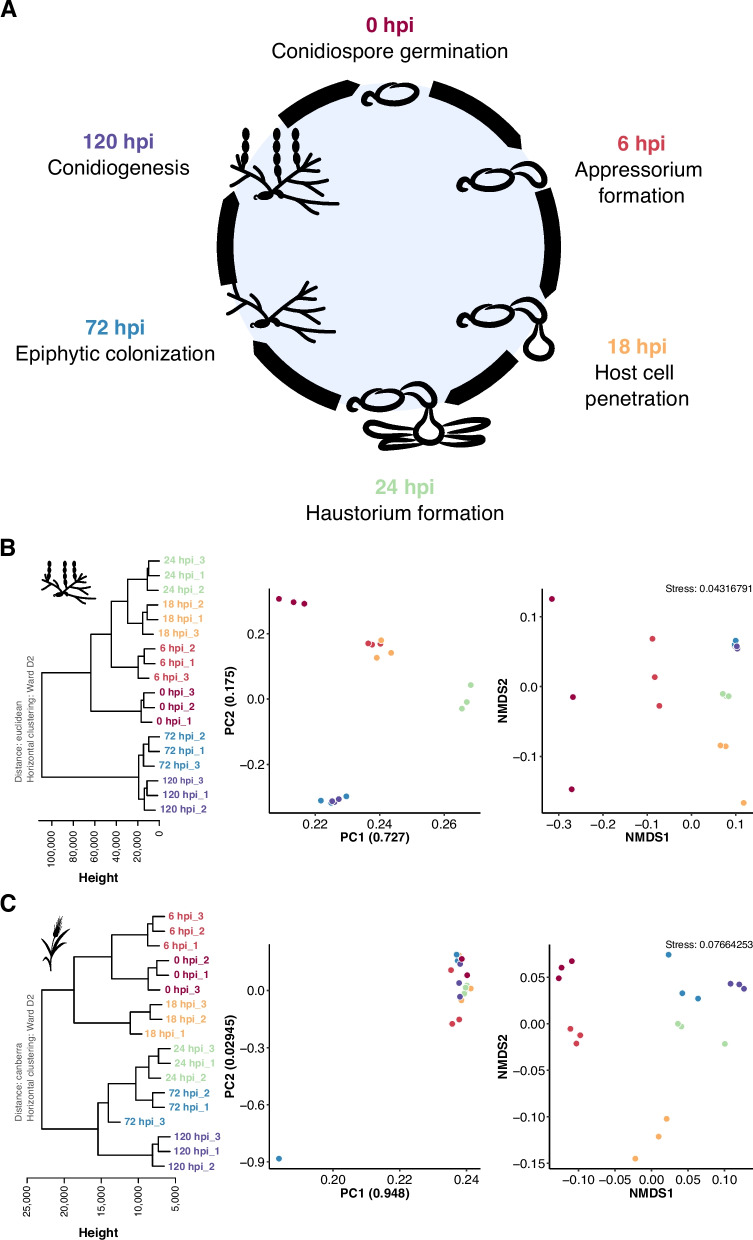
Background: The genome of the obligate biotrophic phytopathogenic barley powdery mildew fungus Blumeria hordei is inflated due to highly abundant and possibly active transposable elements (TEs). In the absence of the otherwise common repeat-induced point mutation transposon defense mechanism, noncoding RNAs could be key for regulating the activity of TEs and coding genes during the pathogenic life cycle.
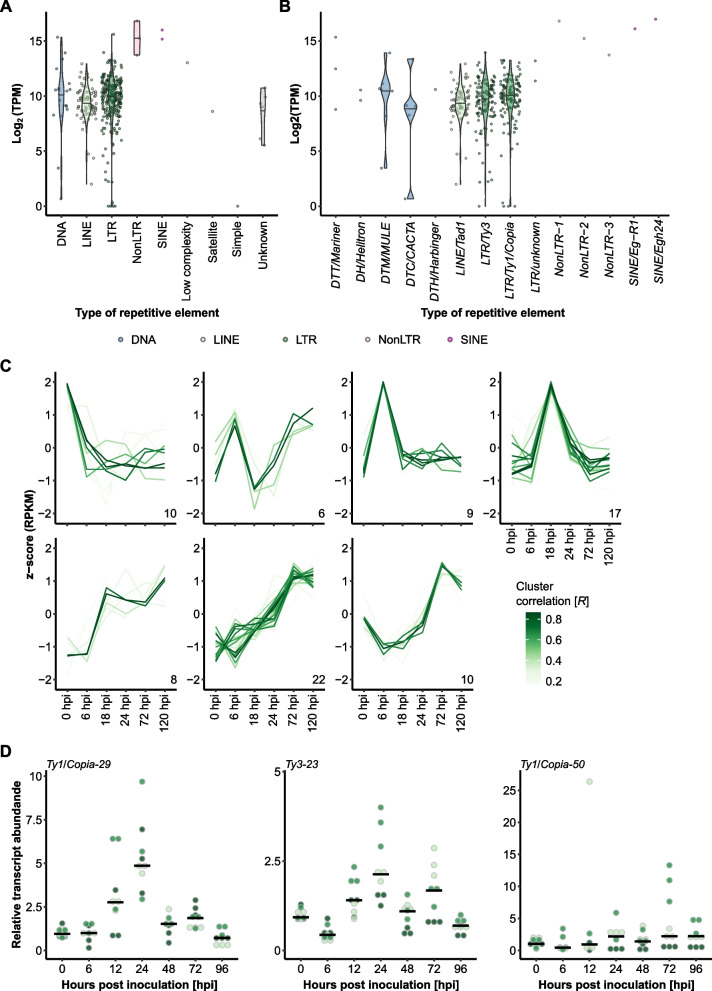
Results: We performed time-course whole-transcriptome shotgun sequencing (RNA-seq) of total RNA derived from infected barley leaf epidermis at various stages of fungal pathogenesis and observed significant transcript accumulation and time point-dependent regulation of TEs in B. hordei. Using a manually curated consensus database of 344 TEs, we discovered phased small RNAs mapping to 104 consensus transposons, suggesting that RNA interference contributes significantly to their regulation. Further, we identified 5,127 long noncoding RNAs (lncRNAs) genome-wide in B. hordei, of which 823 originated from the antisense strand of a TE. Co-expression network analysis of lncRNAs, TEs, and coding genes throughout the asexual life cycle of B. hordei points at extensive positive and negative co-regulation of lncRNAs, subsets of TEs and coding genes.
Conclusions: Our work suggests that similar to mammals and plants, fungal lncRNAs support the dynamic modulation of transcript levels, including TEs, during pivotal stages of host infection. The lncRNAs may support transcriptional diversity and plasticity amid loss of coding genes in powdery mildew fungi and may give rise to novel regulatory elements and virulence peptides, thus representing key drivers of rapid evolutionary adaptation to promote pathogenicity and overcome host defense.
Keywords: Blumeria; Co-expression; Long noncoding RNA (lncRNA); Powdery mildew; RNA interference; Transposable element.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




Similar articles
-
Site-specific analysis reveals candidate cross-kingdom small RNAs, tRNA and rRNA fragments, and signs of fungal RNA phasing in the barley-powdery mildew interaction.Mol Plant Pathol. 2023 Jun;24(6):570-587. doi: 10.1111/mpp.13324. Epub 2023 Mar 14. Mol Plant Pathol. 2023. PMID: 36917011 Free PMC article.
-
Transposable Elements Contribute to the Regulation of Long Noncoding RNAs in Drosophila melanogaster.Insects. 2024 Nov 30;15(12):950. doi: 10.3390/insects15120950. Insects. 2024. PMID: 39769552 Free PMC article.
-
The Parauncinula polyspora Draft Genome Provides Insights into Patterns of Gene Erosion and Genome Expansion in Powdery Mildew Fungi.mBio. 2019 Sep 24;10(5):e01692-19. doi: 10.1128/mBio.01692-19. mBio. 2019. PMID: 31551331 Free PMC article.
-
Transposon-Derived Non-coding RNAs and Their Function in Plants.Front Plant Sci. 2018 May 3;9:600. doi: 10.3389/fpls.2018.00600. eCollection 2018. Front Plant Sci. 2018. PMID: 29774045 Free PMC article. Review.
-
Role of Transposable Elements in Gene Regulation in the Human Genome.Life (Basel). 2021 Feb 4;11(2):118. doi: 10.3390/life11020118. Life (Basel). 2021. PMID: 33557056 Free PMC article. Review.
Cited by
-
Genome analysis of the esca-associated Basidiomycetes Fomitiporia mediterranea, Fomitiporia polymorpha, Inonotus vitis, and Tropicoporus texanus reveals virulence factor repertoires characteristic of white-rot fungi.G3 (Bethesda). 2024 Oct 7;14(10):jkae189. doi: 10.1093/g3journal/jkae189. G3 (Bethesda). 2024. PMID: 39141591 Free PMC article.
-
Mycologists and Virologists Align: Proposing Botrytis cinerea for Global Mycovirus Studies.Viruses. 2024 Sep 18;16(9):1483. doi: 10.3390/v16091483. Viruses. 2024. PMID: 39339959 Free PMC article. Review.
-
Genome-wide identification of long non-coding RNA for Botrytis cinerea during infection to tomato (Solanum lycopersicum) leaves.BMC Genomics. 2025 Jan 6;26(1):7. doi: 10.1186/s12864-024-11171-8. BMC Genomics. 2025. PMID: 39762752 Free PMC article.
-
Exploring the hidden hot world of long non-coding RNAs in thermophilic fungus using a robust computational pipeline.Sci Rep. 2024 Aug 27;14(1):19797. doi: 10.1038/s41598-024-67975-x. Sci Rep. 2024. PMID: 39187522 Free PMC article.
-
A powdery mildew core effector protein targets the host endosome tethering complexes HOPS and CORVET in barley.Plant Physiol. 2025 Mar 28;197(4):kiaf067. doi: 10.1093/plphys/kiaf067. Plant Physiol. 2025. PMID: 39973312 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
