

Machine learning for microbiologists
- PMID: 37968359
- PMCID: PMC11980903
- DOI: 10.1038/s41579-023-00984-1
Machine learning for microbiologists
Abstract
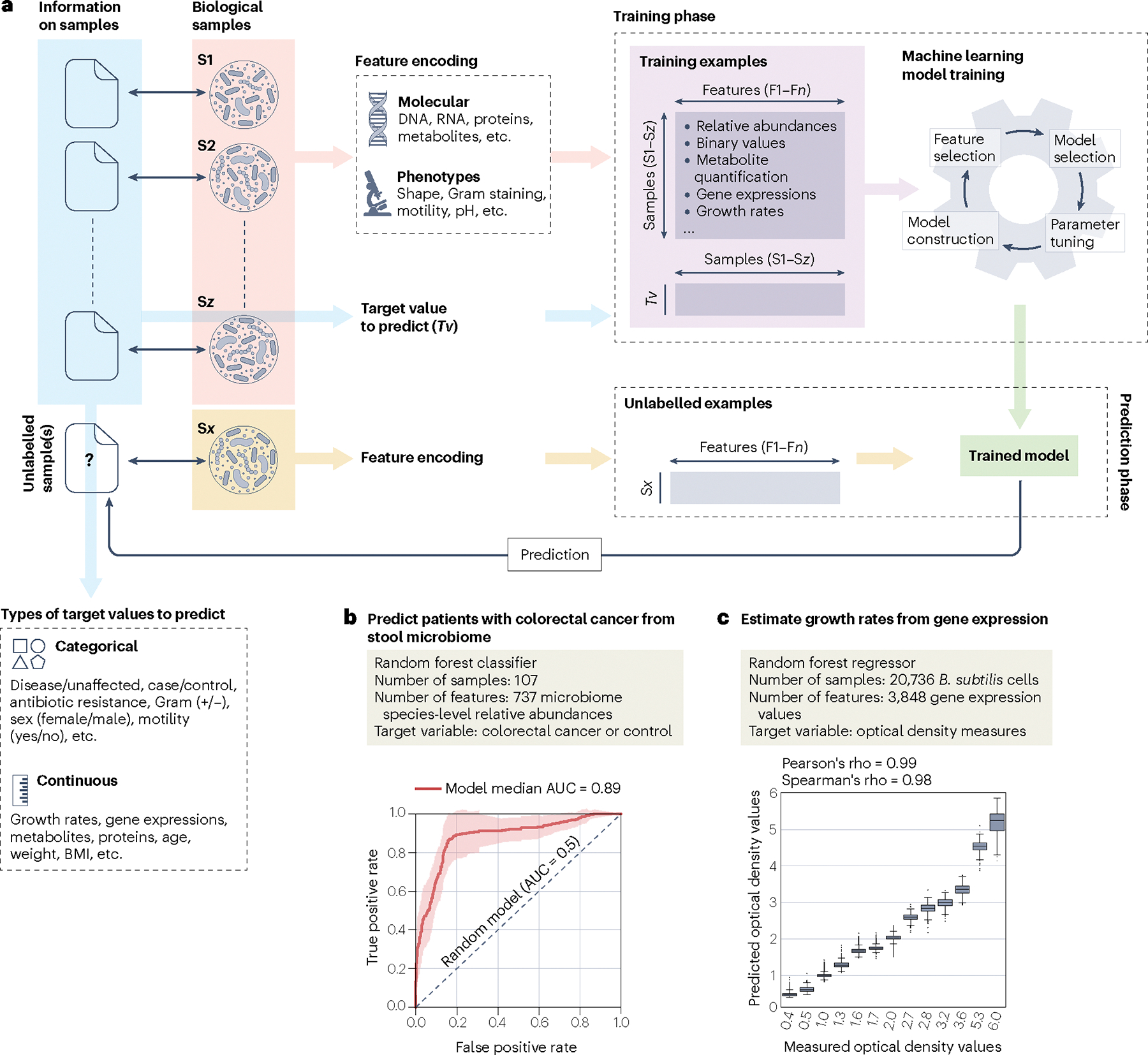
Machine learning is increasingly important in microbiology where it is used for tasks such as predicting antibiotic resistance and associating human microbiome features with complex host diseases. The applications in microbiology are quickly expanding and the machine learning tools frequently used in basic and clinical research range from classification and regression to clustering and dimensionality reduction. In this Review, we examine the main machine learning concepts, tasks and applications that are relevant for experimental and clinical microbiologists. We provide the minimal toolbox for a microbiologist to be able to understand, interpret and use machine learning in their experimental and translational activities.
© 2023. Springer Nature Limited.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures



References
-
- Bishop CM Pattern recognition and machine learning (Springer, 2006).
-
- Hastie T, Tibshirani R & Friedman J The Elements of Statistical Learning: Data Mining, Inference, and Prediction 2nd edn (Springer Science & Business Media, 2009).
-
- James G, Witten D, Hastie T & Tibshirani R An Introduction to Statistical Learning: with Applications in R (Springer Science & Business Media, 2013).
-
- Murphy KP Probabilistic Machine Learning: Advanced Topics (MIT Press, 2022).
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
