

Clonal selection of hematopoietic stem cells after gene therapy for sickle cell disease
- PMID: 37973947
- PMCID: PMC10719109
- DOI: 10.1038/s41591-023-02636-6
Clonal selection of hematopoietic stem cells after gene therapy for sickle cell disease
Abstract
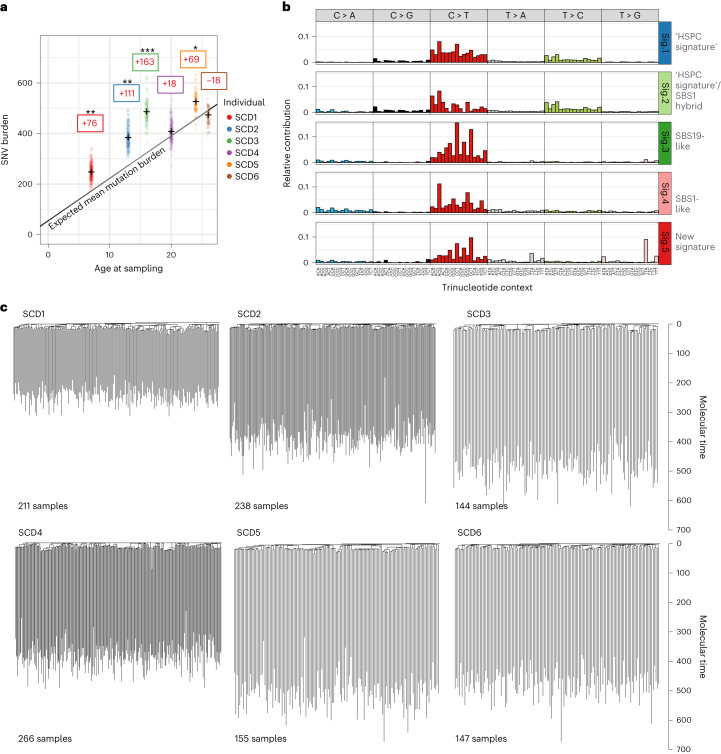
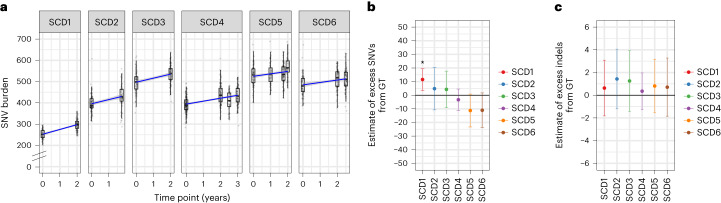
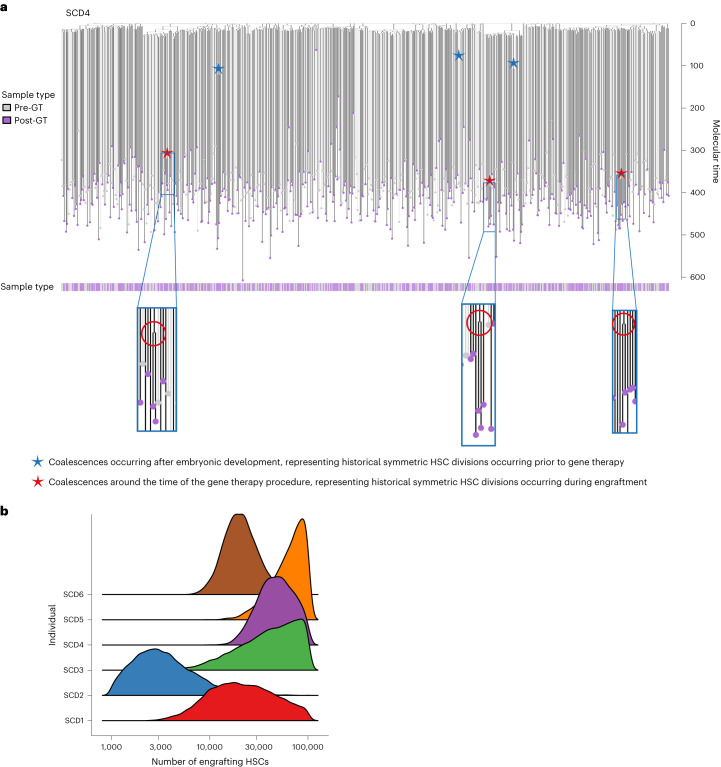
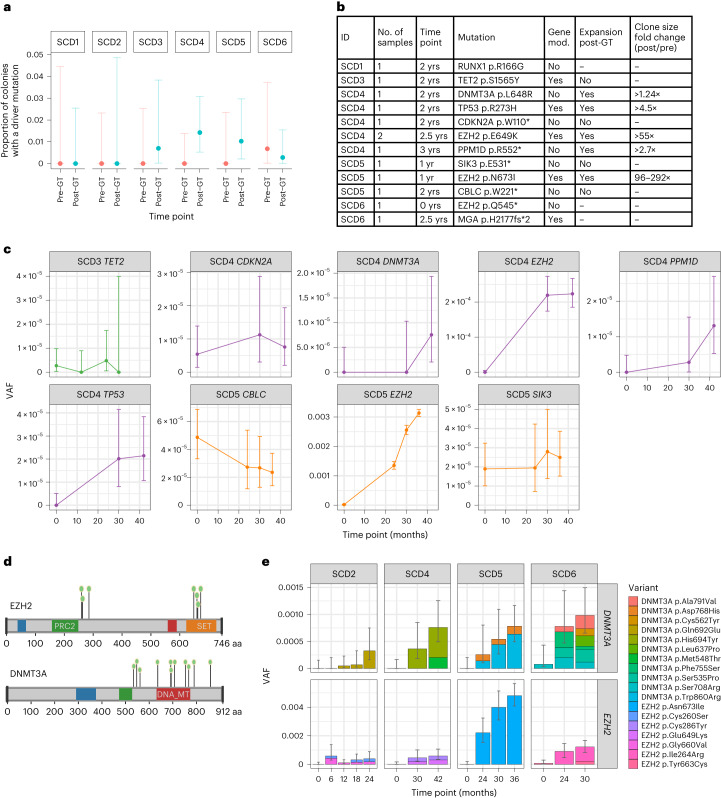
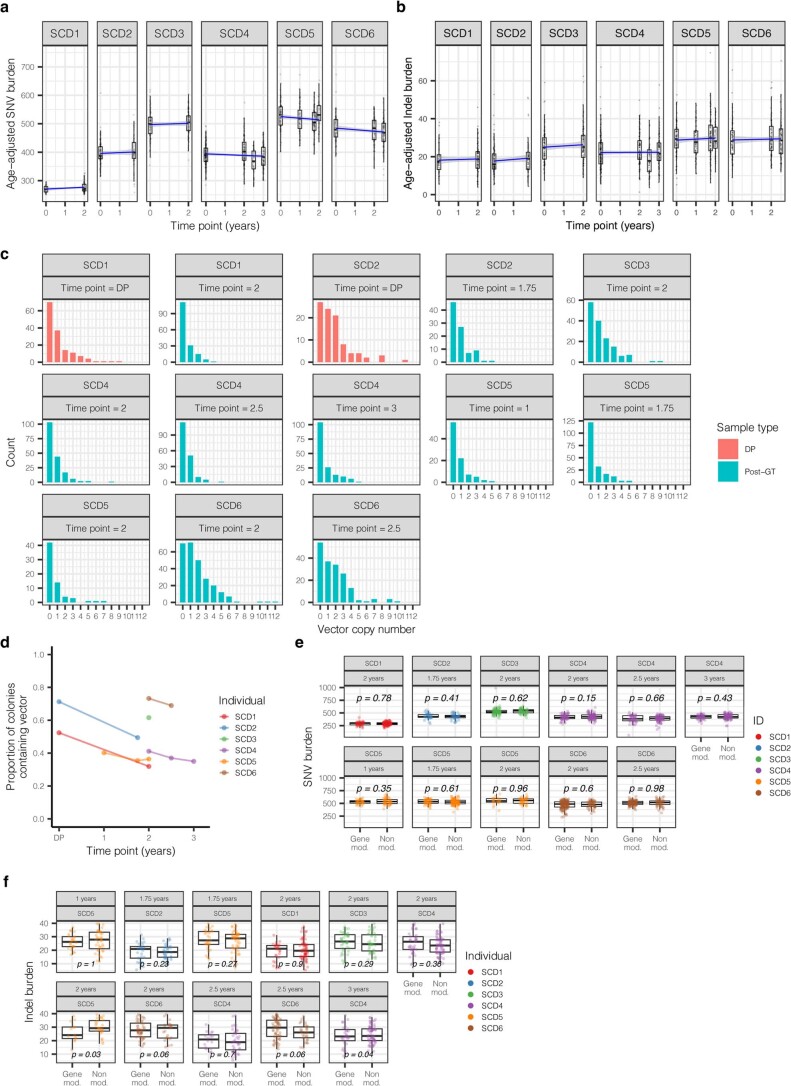
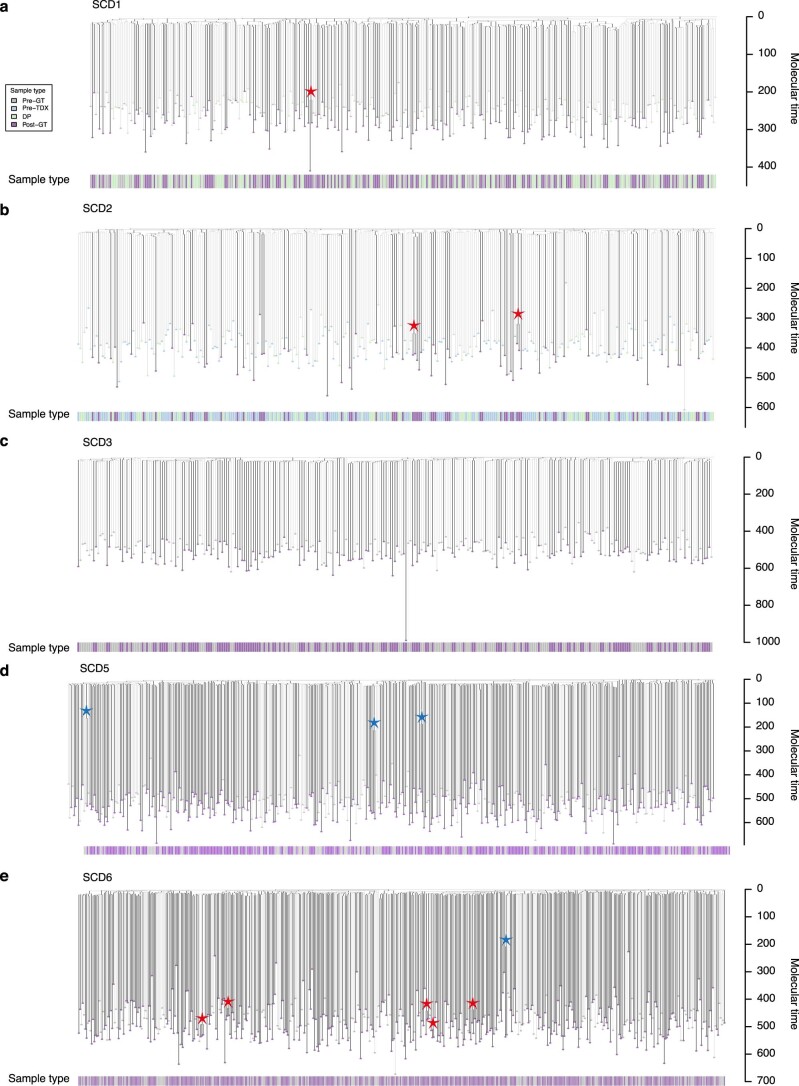
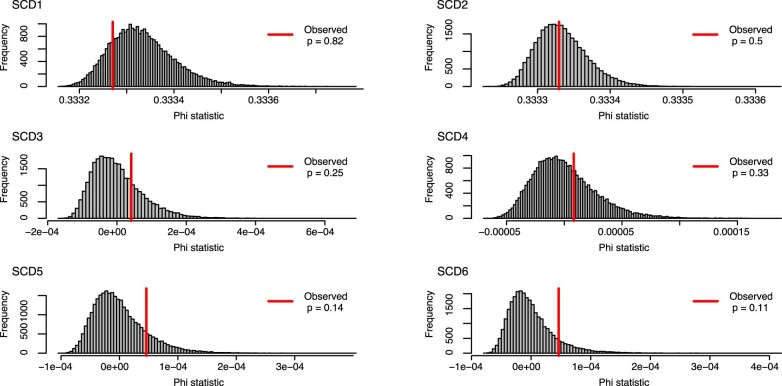
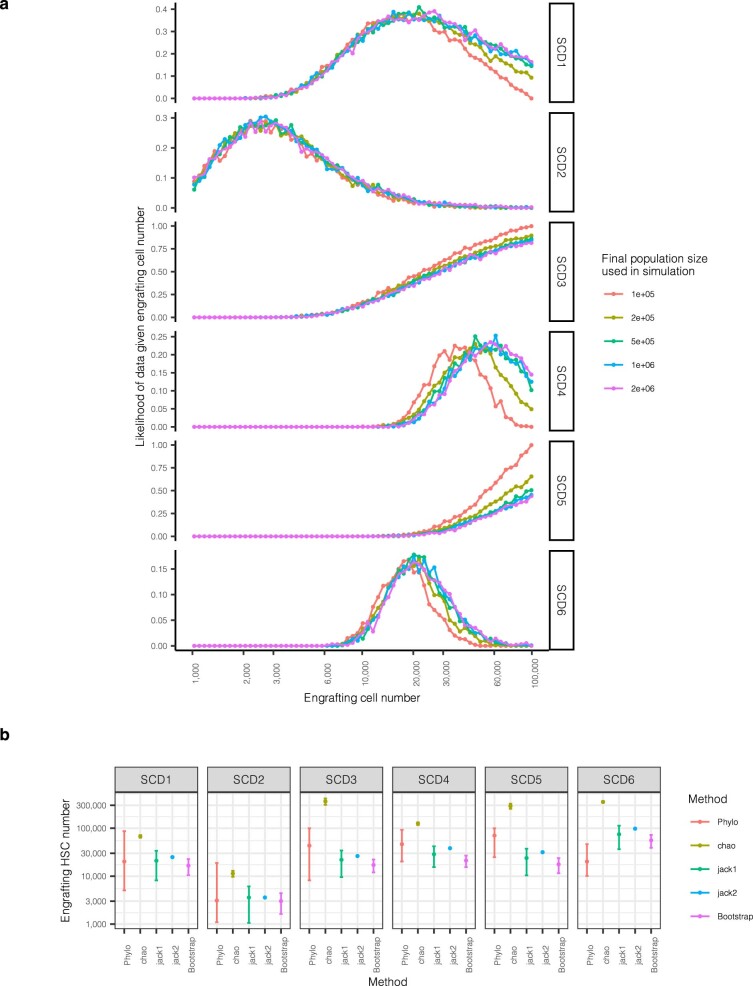
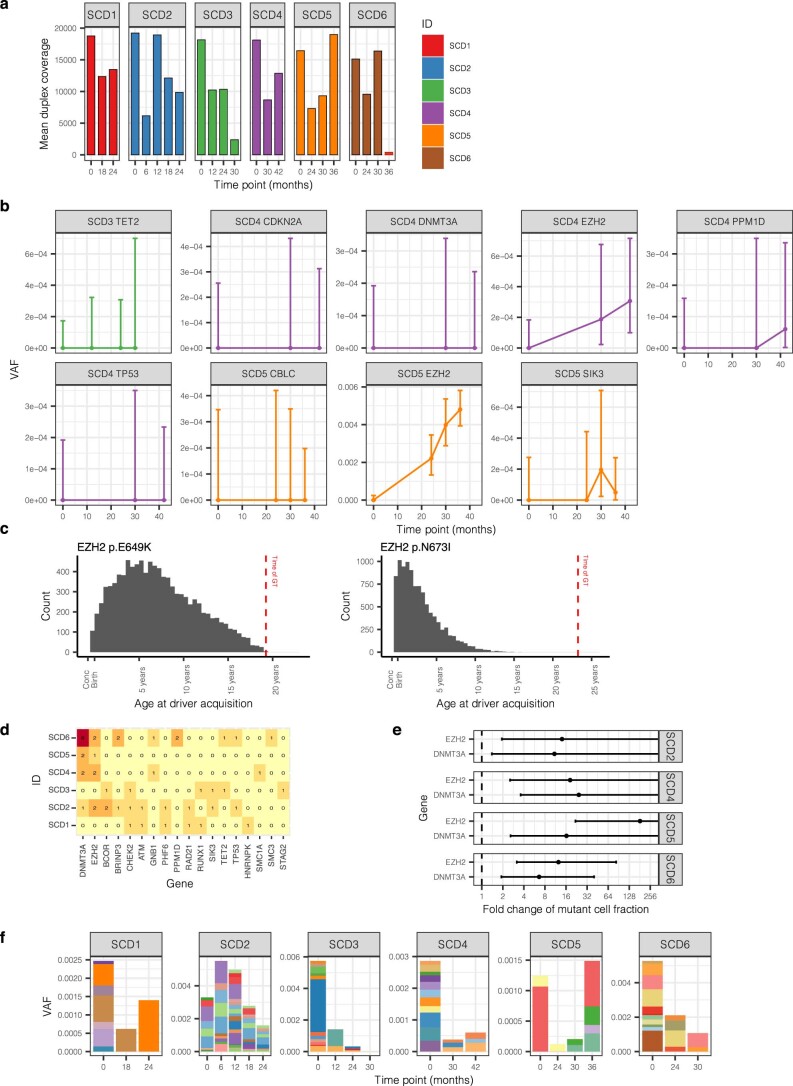
Gene therapy (GT) provides a potentially curative treatment option for patients with sickle cell disease (SCD); however, the occurrence of myeloid malignancies in GT clinical trials has prompted concern, with several postulated mechanisms. Here, we used whole-genome sequencing to track hematopoietic stem cells (HSCs) from six patients with SCD at pre- and post-GT time points to map the somatic mutation and clonal landscape of gene-modified and unmodified HSCs. Pre-GT, phylogenetic trees were highly polyclonal and mutation burdens per cell were elevated in some, but not all, patients. Post-GT, no clonal expansions were identified among gene-modified or unmodified cells; however, an increased frequency of potential driver mutations associated with myeloid neoplasms or clonal hematopoiesis (DNMT3A- and EZH2-mutated clones in particular) was observed in both genetically modified and unmodified cells, suggesting positive selection of mutant clones during GT. This work sheds light on HSC clonal dynamics and the mutational landscape after GT in SCD, highlighting the enhanced fitness of some HSCs harboring pre-existing driver mutations. Future studies should define the long-term fate of mutant clones, including any contribution to expansions associated with myeloid neoplasms.
© 2023. The Author(s).
Conflict of interest statement
P.J.C. is a co-founder, stockholder and consultant for FL86. M.A.F. is an employee and stockholder of AstraZeneca. D.G.K. receives laboratory funding from STRM.bio. D.A.W. serves on the following committees: Novartis steering committee, Beam Therapeutics scientific advisory board, Skyline Therapeutics (formerly Geneception) scientific advisory board and Biomarin insertion site advisor board. D.A.W. also acts as a consultant for Verve Therapeutics and Monte Rosa Therapeutics. He also has research funding from ExCellThera. E.B.E. served as a consultant for a bluebird bio steering committee. The remaining authors declare no competing interests.
Figures
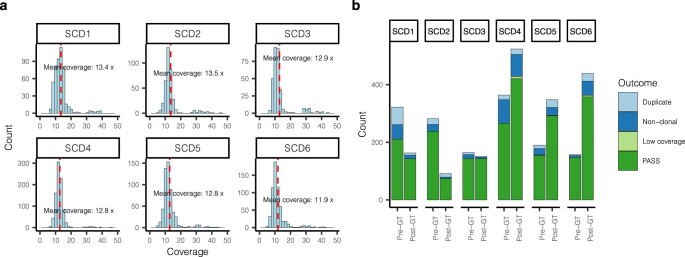
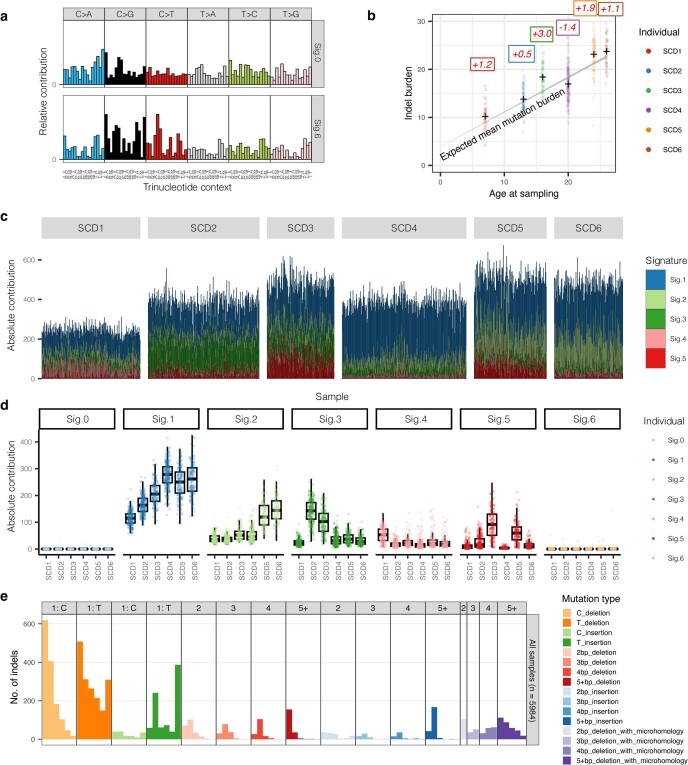
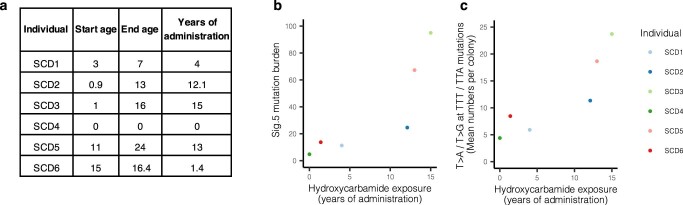
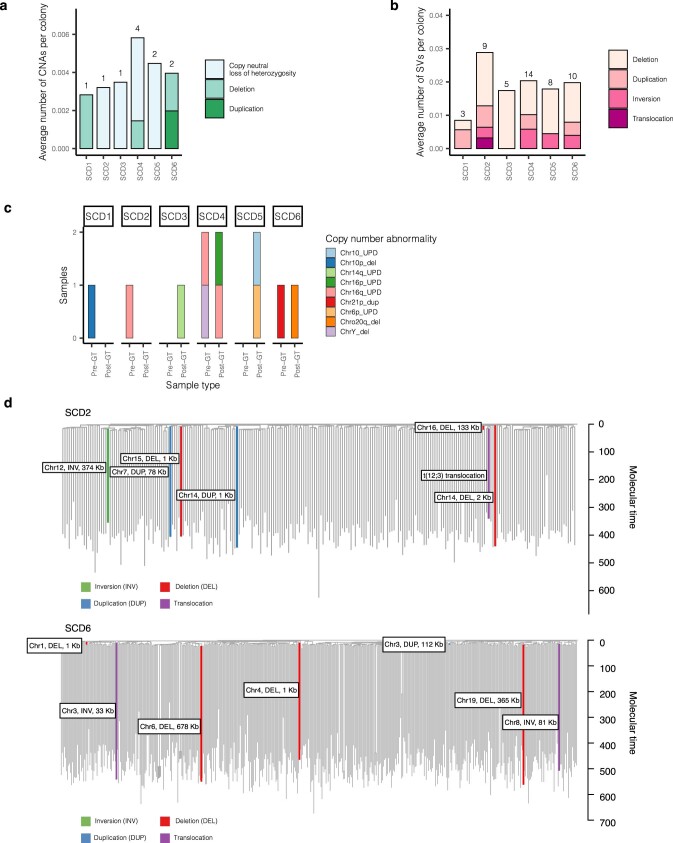

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
