

Variational autoencoder-based chemical latent space for large molecular structures with 3D complexity
- PMID: 37973971
- PMCID: PMC10654724
- DOI: 10.1038/s42004-023-01054-6
Variational autoencoder-based chemical latent space for large molecular structures with 3D complexity
Abstract
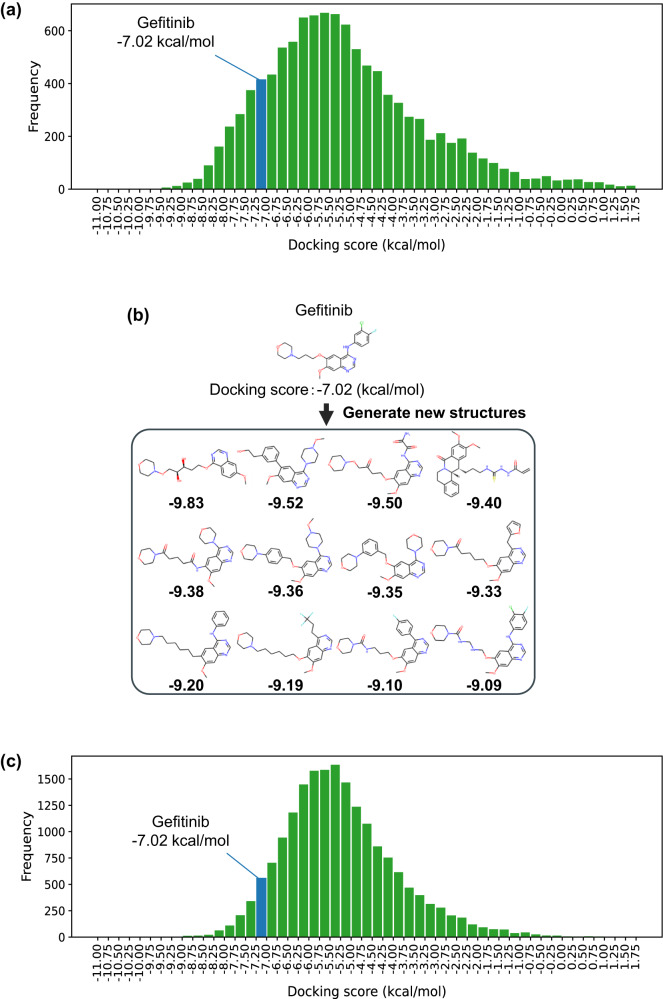
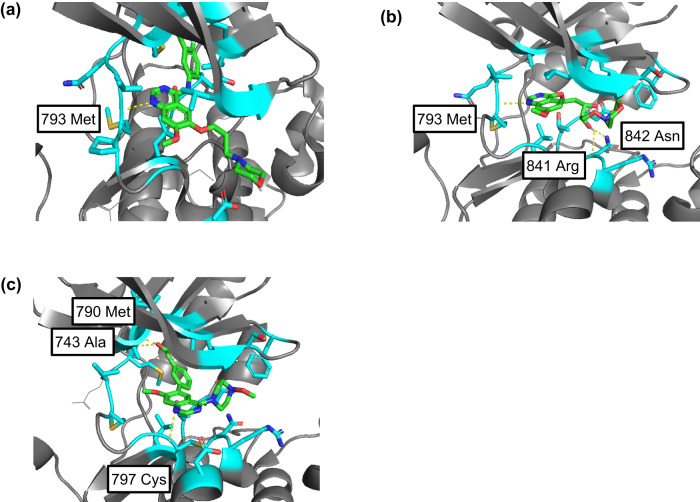
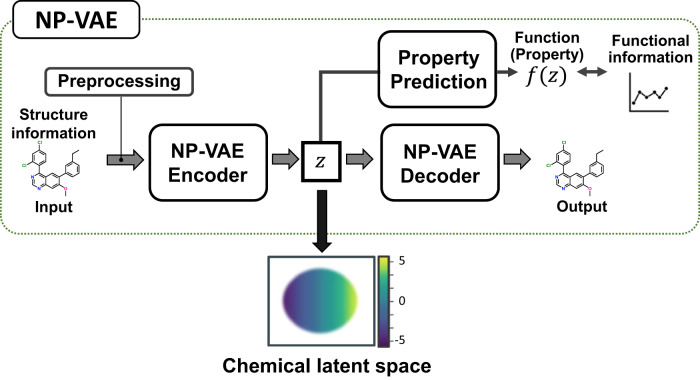
The structural diversity of chemical libraries, which are systematic collections of compounds that have potential to bind to biomolecules, can be represented by chemical latent space. A chemical latent space is a projection of a compound structure into a mathematical space based on several molecular features, and it can express structural diversity within a compound library in order to explore a broader chemical space and generate novel compound structures for drug candidates. In this study, we developed a deep-learning method, called NP-VAE (Natural Product-oriented Variational Autoencoder), based on variational autoencoder for managing hard-to-analyze datasets from DrugBank and large molecular structures such as natural compounds with chirality, an essential factor in the 3D complexity of compounds. NP-VAE was successful in constructing the chemical latent space from large-sized compounds that were unable to be handled in existing methods, achieving higher reconstruction accuracy, and demonstrating stable performance as a generative model across various indices. Furthermore, by exploring the acquired latent space, we succeeded in comprehensively analyzing a compound library containing natural compounds and generating novel compound structures with optimized functions.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
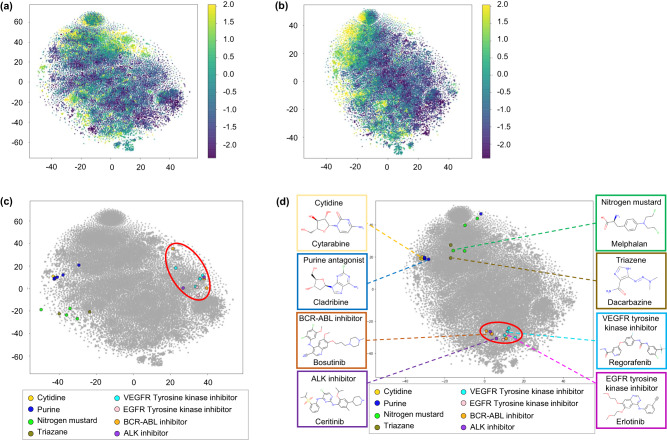
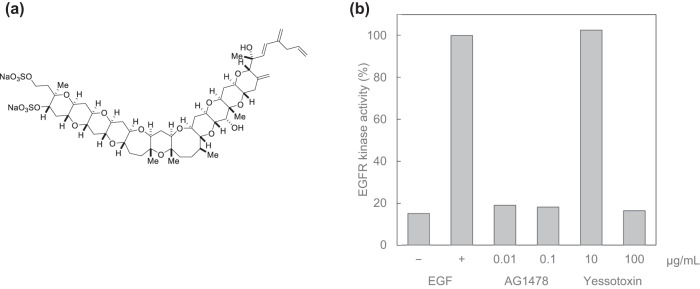
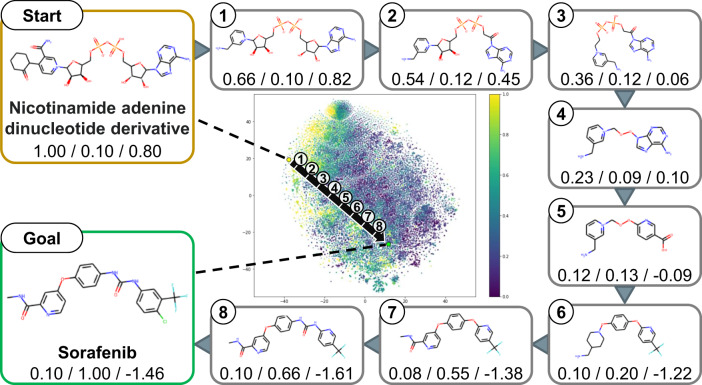
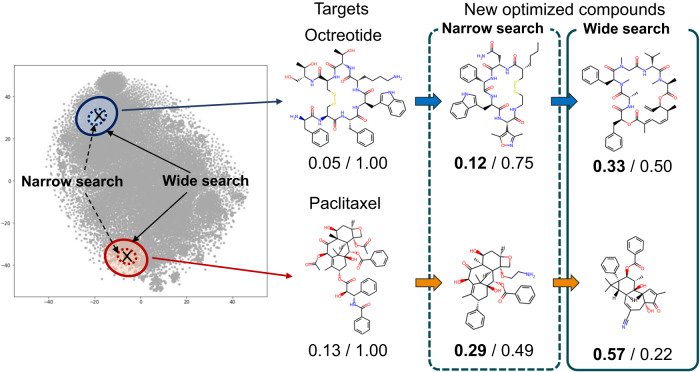
References
-
- Kingma D. P., Welling M. Auto-encoding variational Bayes. Preprint at https://arxiv.org/abs/1312.6114 (2013).
Grants and funding
- 22H04901/Ministry of Education, Culture, Sports, Science and Technology (MEXT)
- 17H06410/Ministry of Education, Culture, Sports, Science and Technology (MEXT)
- 23H04885/Ministry of Education, Culture, Sports, Science and Technology (MEXT)
- 23H04880/Ministry of Education, Culture, Sports, Science and Technology (MEXT)
- 23H04881/Ministry of Education, Culture, Sports, Science and Technology (MEXT)
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
