

The phylogeny of Acetobacteraceae: photosynthetic traits and deranged respiratory enzymes
- PMID: 37975678
- PMCID: PMC10715153
- DOI: 10.1128/spectrum.00575-23
The phylogeny of Acetobacteraceae: photosynthetic traits and deranged respiratory enzymes
Abstract
Acetobacteraceae are one of the best known and most extensively studied groups of bacteria, which nowadays encompasses a variety of taxa that are very different from the vinegar-producing species defining the family. Our paper presents the most detailed phylogeny of all current taxa classified as Acetobacteraceae, for which we propose a taxonomic revision. Several of such taxa inhabit some of the most extreme environments on the planet, from the deserts of Antarctica to the Sinai desert, as well as acidic niches in volcanic sites like the one we have been studying in Patagonia. Our work documents the progressive variation of the respiratory chain in early branching Acetobacteraceae into the different respiratory chains of acidophilic taxa such as Acidocella and acetous taxa such as Acetobacter. Remarkably, several genomes retain remnants of ancestral photosynthetic traits and functional bc 1 complexes. Thus, we propose that the common ancestor of Acetobacteraceae was photosynthetic.
Keywords: Acetobacteraceae; bacterial phylogeny; energy metabolism; phylogenomics.
Conflict of interest statement
The authors declare no conflict of interest.
Figures




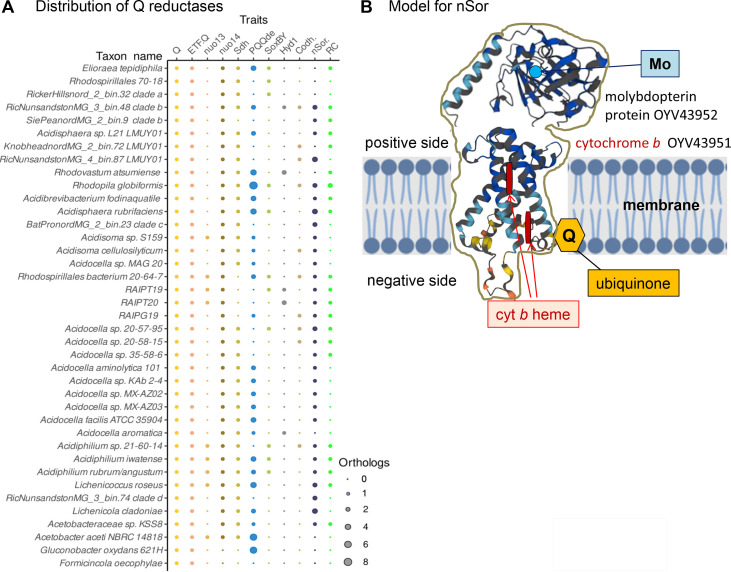
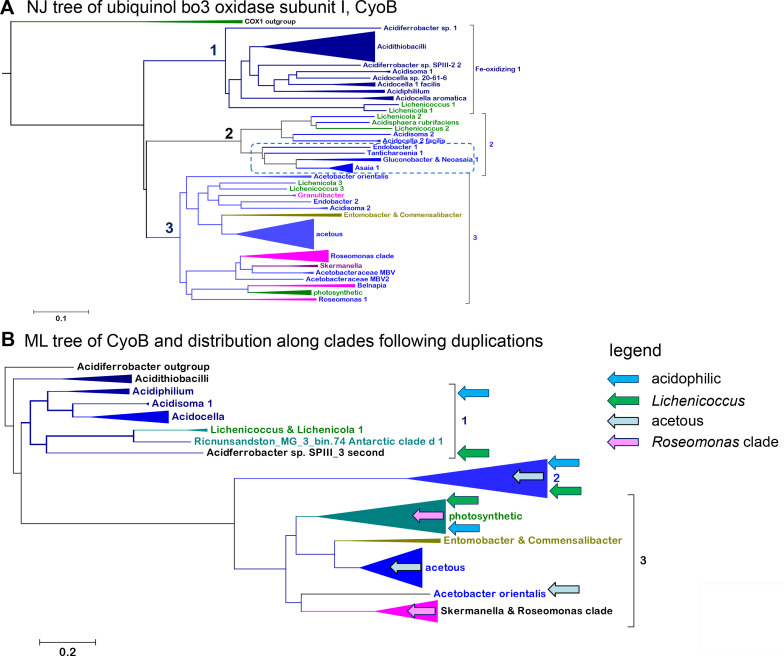


References
-
- Komagata K, Lino T, Yamada Y. 2014. The family Acetobacteraceae, p 3–78. In Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F (ed), The prokaryotes: alphaproteobacteria and betaproteobacteria. Springer Berlin Heidelberg.
-
- Hördt A, López MG, Meier-Kolthoff JP, Schleuning M, Weinhold LM, Tindall BJ, Gronow S, Kyrpides NC, Woyke T, Göker M. 2020. Analysis of 1,000+ type-strain genomes substantially improves taxonomic classification of Alphaproteobacteria Front Microbiol 11:468. doi:10.3389/fmicb.2020.00468 - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
