

Autophagy activates EGR1 via MAPK/ERK to induce FGF2 in renal tubular cells for fibroblast activation and fibrosis during maladaptive kidney repair
- PMID: 37978868
- PMCID: PMC11135847
- DOI: 10.1080/15548627.2023.2281156
Autophagy activates EGR1 via MAPK/ERK to induce FGF2 in renal tubular cells for fibroblast activation and fibrosis during maladaptive kidney repair
Abstract
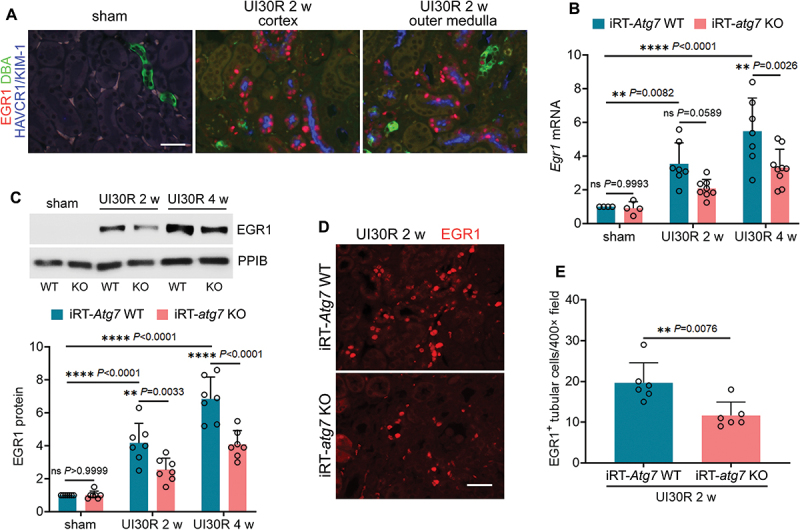
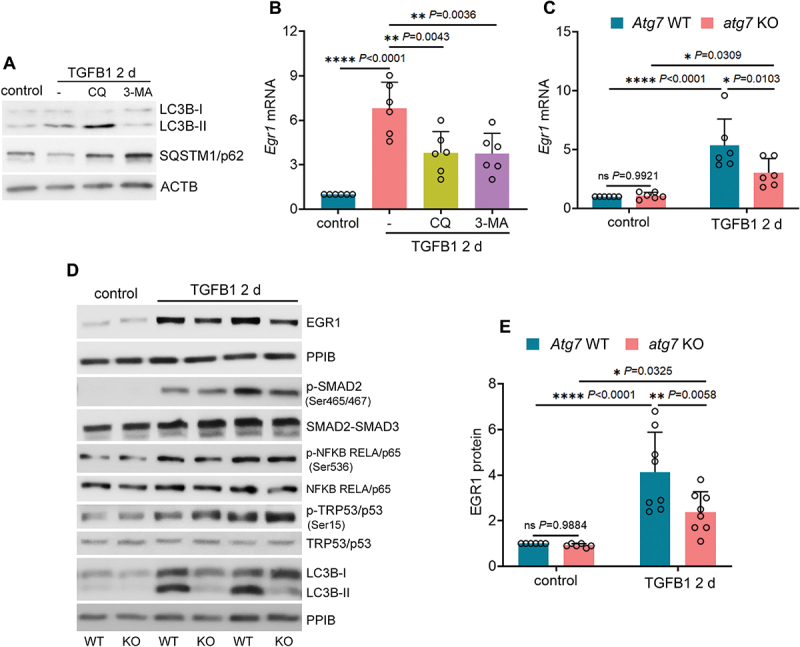
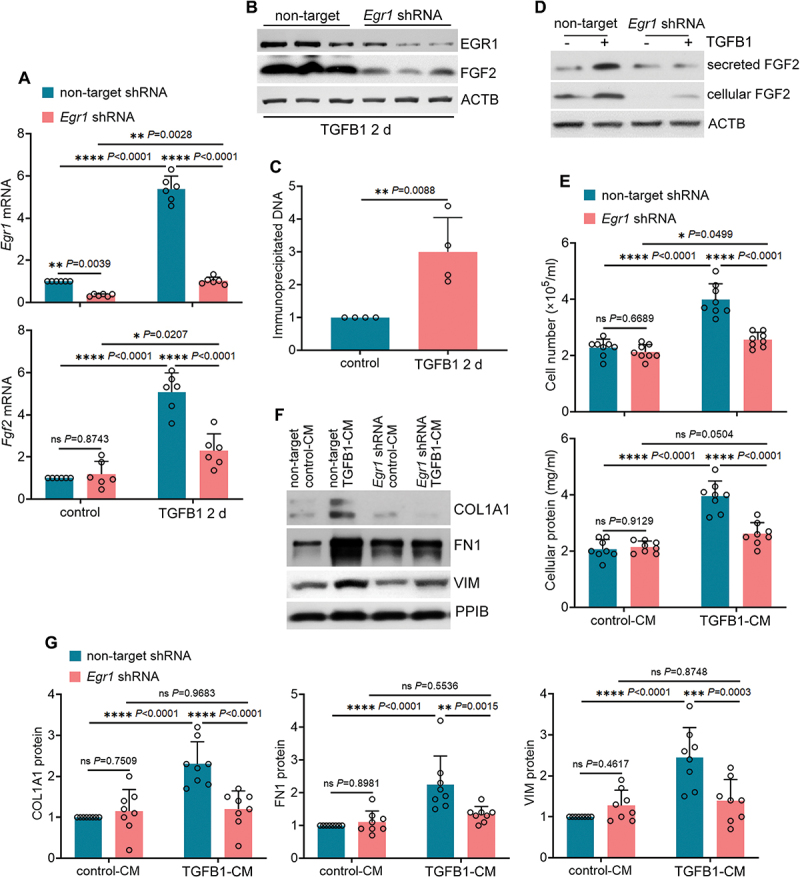
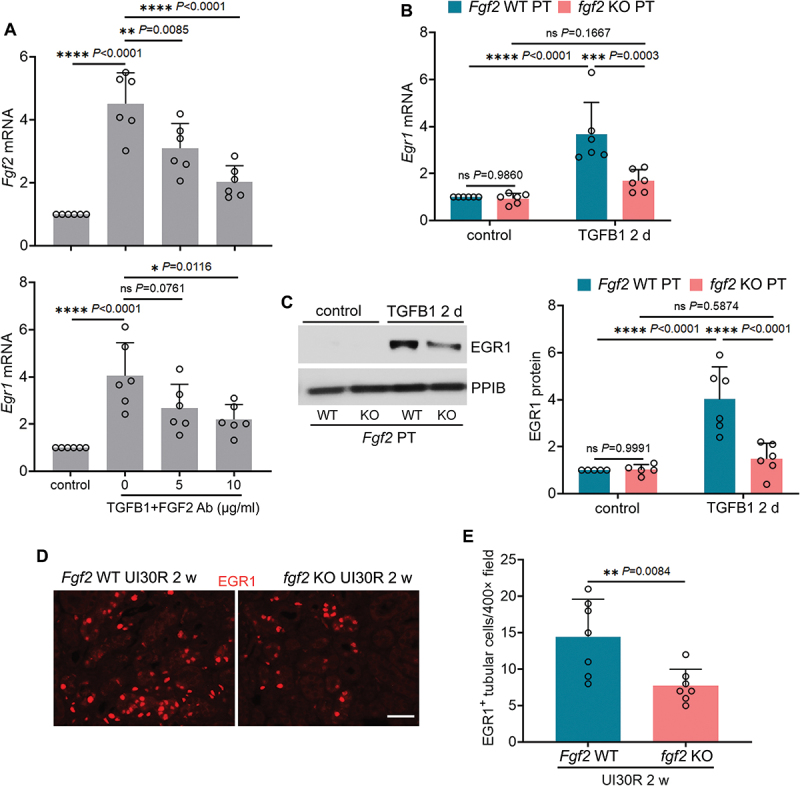
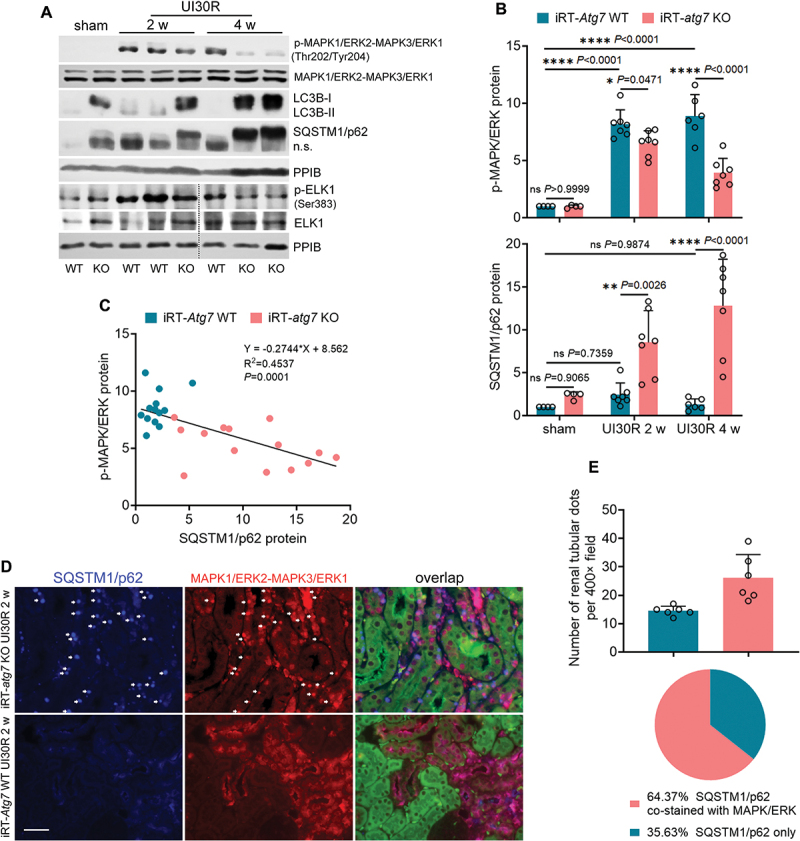
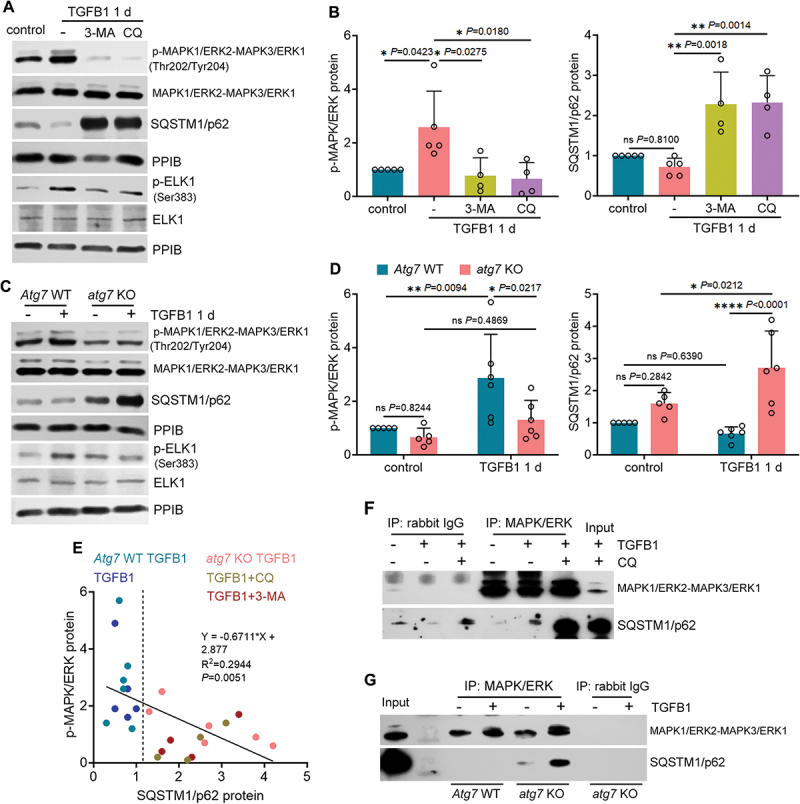
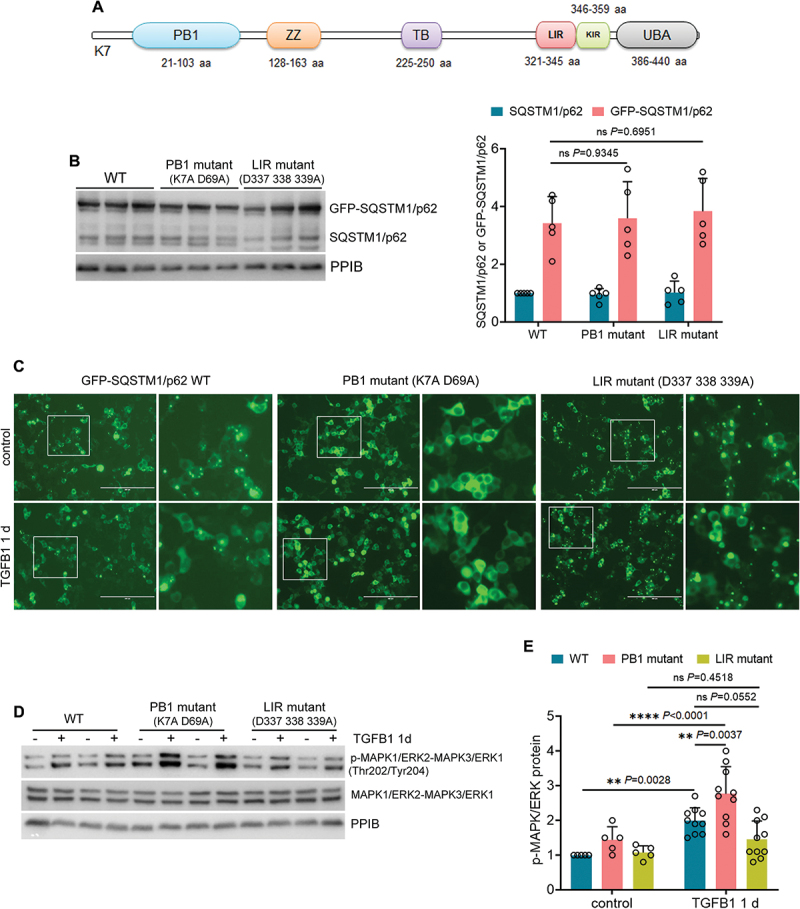
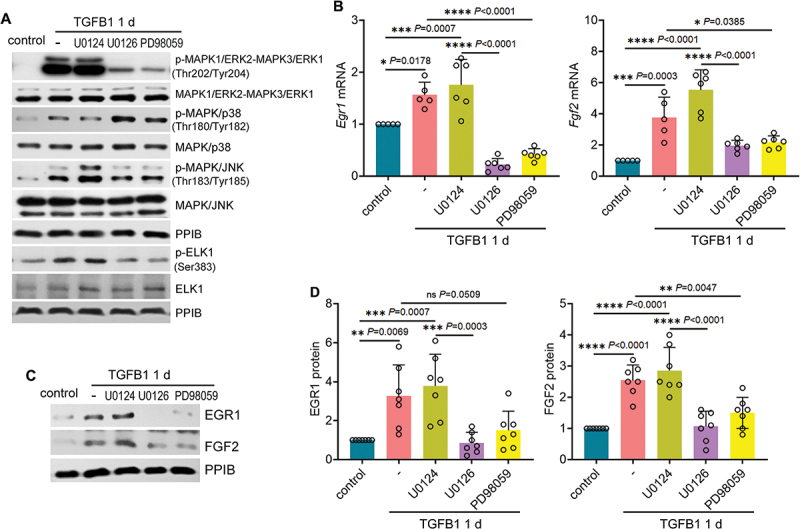
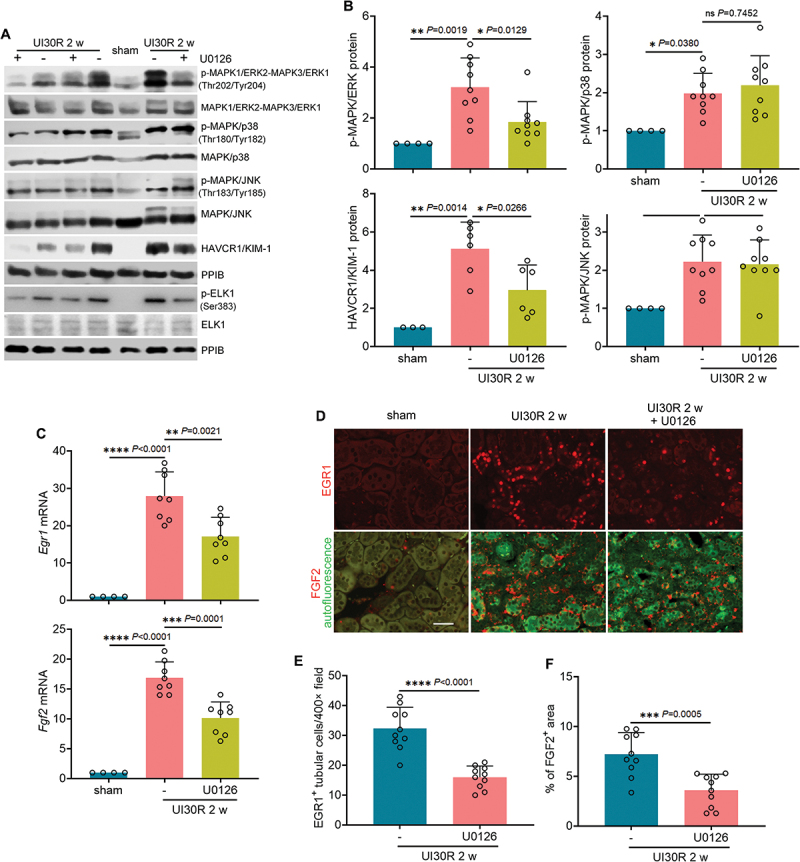
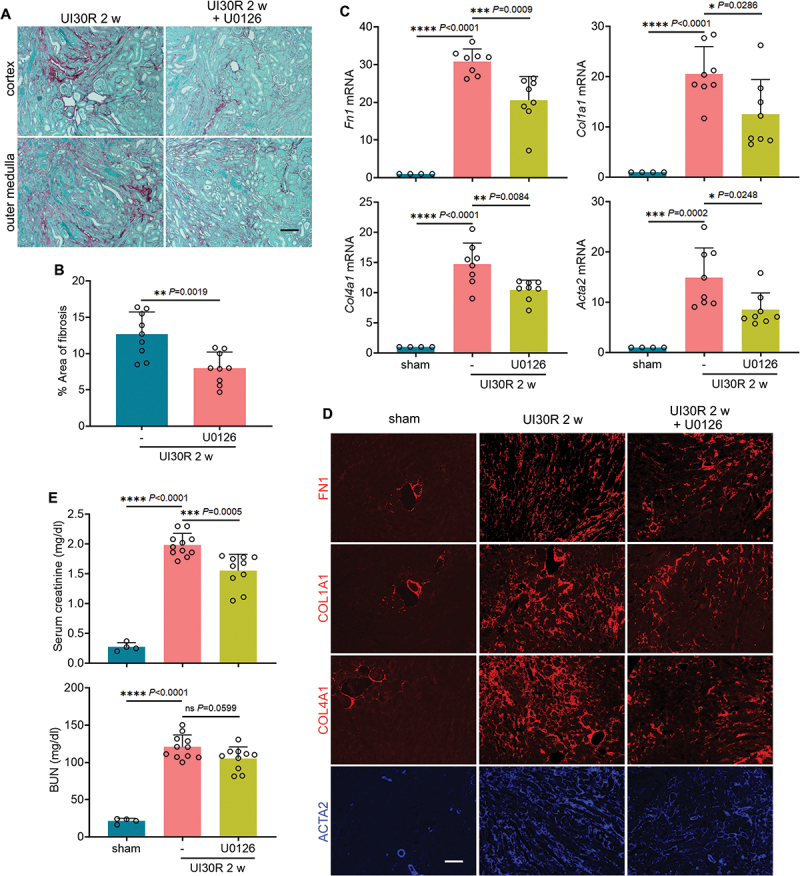
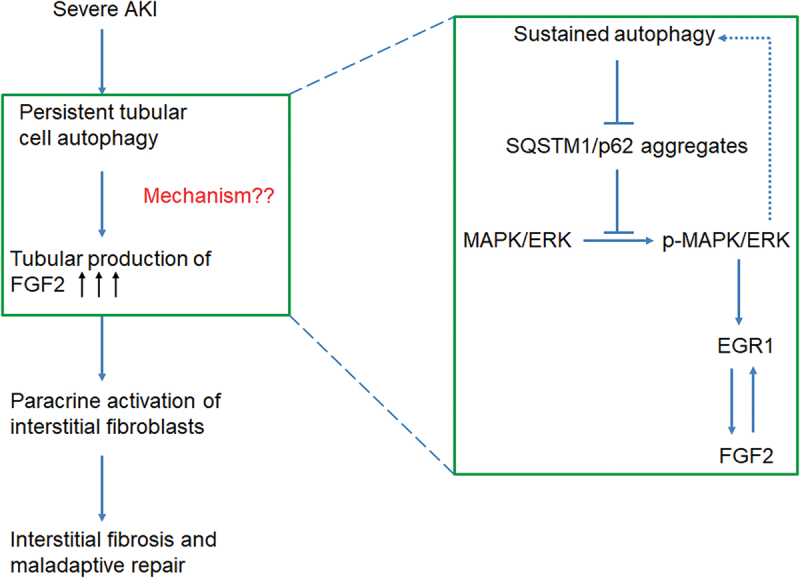
Macroautophagy/autophagy contributes to maladaptive kidney repair by inducing pro-fibrotic factors such as FGF2 (fibroblast growth factor 2), but the underlying mechanism remains elusive. Here, we show that EGR1 (early growth response 1) was induced in injured proximal tubules after ischemic acute kidney injury (AKI) and this induction was suppressed by autophagy deficiency in inducible, renal tubule-specific atg7 (autophagy related 7) knockout (iRT-atg7 KO) mice. In cultured proximal tubular cells, TGFB1 (transforming growth factor beta 1) induced EGR1 and this induction was also autophagy dependent. Egr1 knockdown in tubular cells reduced FGF2 expression during TGFB1 treatment, leading to less FGF2 secretion and decreased paracrine effects on fibroblasts. ChIP assay detected an increased binding of EGR1 to the Fgf2 gene promoter in TGFB1-treated tubular cells. Both Fgf2 and Egr1 transcription was inhibited by FGF2 neutralizing antibody, suggesting a positive feedback for EGR1-mediated FGF2 autoregulation. This feedback was confirmed using fgf2-deficient tubular cells and fgf2-deficient mice. Upstream of EGR1, autophagy deficiency in mice suppressed MAPK/ERK (mitogen-activated protein kinase) activation in post-ischemic renal tubules. This inhibition correlated with SQSTM1/p62 (sequestosome 1) aggregation and its sequestration of MAPK/ERK. SQSTM1/p62 interacted with MAPK/ERK and blocked its activation during TGFB1 treatment in autophagy-deficient tubular cells. Inhibition of MAPK/ERK suppressed EGR1 and FGF2 expression in maladaptive tubules, leading to the amelioration of renal fibrosis and improvement of renal function. These results suggest that autophagy activates MAPK/ERK in renal tubular cells, which induces EGR1 to transactivate FGF2. FGF2 is then secreted into the interstitium to stimulate fibroblasts for fibrogenesis.Abbreviation: 3-MA: 3-methyladenine; ACTA2/α-SMA: actin alpha 2, smooth muscle, aorta; ACTB/β-actin: actin, beta; AKI: acute kidney injury; aa: amino acid; ATG/Atg: autophagy related; BUN: blood urea nitrogen; ChIP: chromatin immunoprecipitation; CKD: chronic kidney disease; CM: conditioned medium; COL1A1: collagen, type I, alpha 1; COL4A1: collagen, type IV, alpha 1; CQ: chloroquine; DBA: dolichos biflorus agglutinin; EGR1: early growth response 1; ELK1: ELK1, member of ETS oncogene family; FGF2: fibroblast growth factor 2; FN1: fibronectin 1; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; HAVCR1/KIM-1: hepatitis A virus cellular receptor 1; IP: immunoprecipitation; LIR: LC3-interacting region; MAP1LC3B/LC3B: microtubule-associated protein 1 light chain 3 beta; MAP2K/MEK: mitogen-activated protein kinase kinase; MAPK: mitogen-activated protein kinase; NFKB: nuclear factor kappa B; PB1: Phox and Bem1; PFT: pifithrin α; PPIB/cyclophilin B: peptidylprolyl isomerase B; RT-qPCR: real time-quantitative PCR; SQSTM1/p62: sequestosome 1; TGFB1/TGF-β1: transforming growth factor beta 1; VIM: vimentin.
Keywords: Interstitial fibrosis; SQSTM1/p62; ischemic acute kidney injury; kidney repair; tubulo-interstitial communication.
Conflict of interest statement
No potential conflict of interest was reported by the author(s).
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous