

Germline bi-allelic SH2B3/LNK alteration predisposes to a neonatal juvenile myelomonocytic leukemia-like disorder
- PMID: 37981895
- PMCID: PMC11290538
- DOI: 10.3324/haematol.2023.283917
Germline bi-allelic SH2B3/LNK alteration predisposes to a neonatal juvenile myelomonocytic leukemia-like disorder
Abstract
Juvenile myelomonocytic leukemia (JMML) is a rare, generally aggressive myeloproliferative neoplasm affecting young children. It is characterized by granulomonocytic expansion, with monocytosis infiltrating peripheral tissues. JMML is initiated by mutations upregulating RAS signaling. Approximately 10% of cases remain without an identified driver event. Exome sequencing of two unrelated cases of familial JMML of unknown genetics and analysis of the French JMML cohort identified 11 patients with variants in SH2B3, encoding LNK, a negative regulator of the JAK-STAT pathway. All variants were absent from healthy population databases, and the mutation spectrum was consistent with a loss of function of the LNK protein. A stoploss variant was shown to affect both protein synthesis and stability. The other variants were either truncating or missense, the latter affecting the SH2 domain that interacts with activated JAK. Of the 11 patients, eight from five families inherited pathogenic bi-allelic SH2B3 germline variants from their unaffected heterozygous parents. These children represent half of the cases with no identified causal mutation in the French cohort. They displayed typical clinical and hematologic features of JMML with neonatal onset and marked thrombocytopenia. They had a hypomethylated DNA profile with fetal characteristics and did not have additional genetic alterations. All patients showed partial or complete spontaneous clinical resolution. However, progression to thrombocythemia and immunity-related pathologies may be of concern later in life. Bi-allelic SH2B3 germline mutations thus define a new condition predisposing to a JMML-like disorder, suggesting that JAK pathway deregulation is capable of initiating JMML, and opening new therapeutic options.
Figures
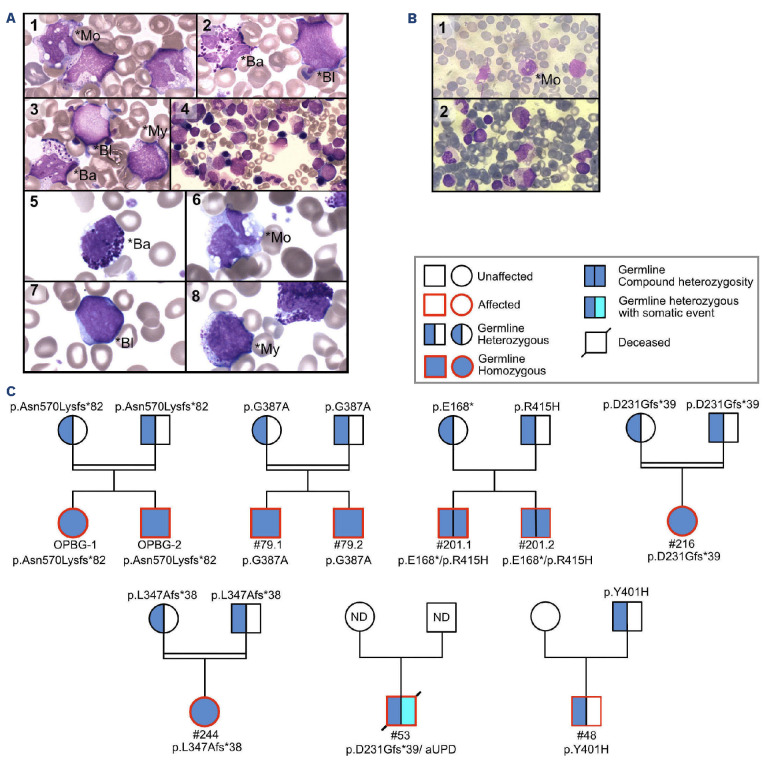
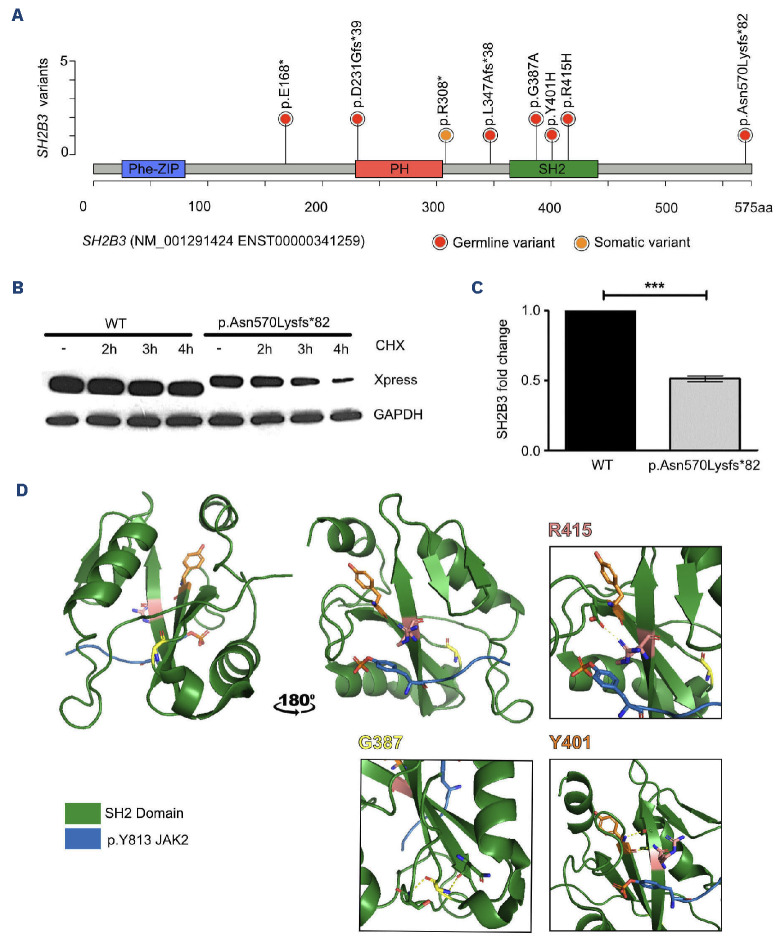
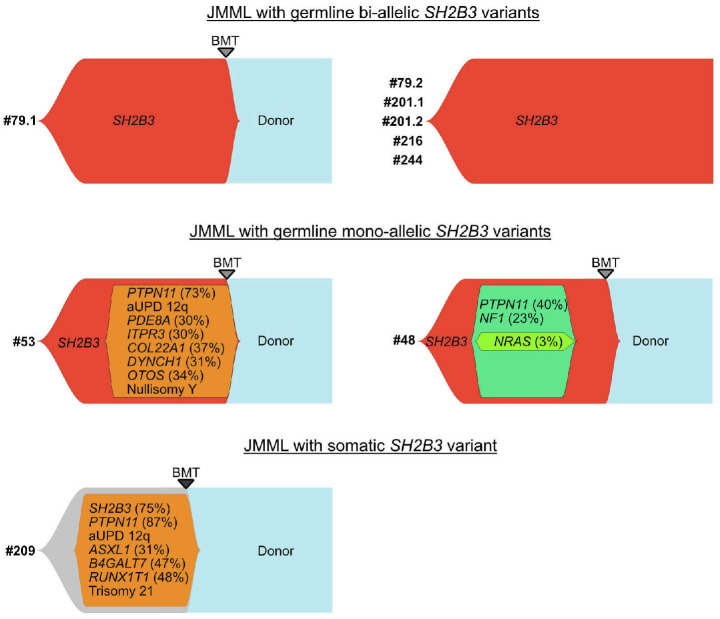
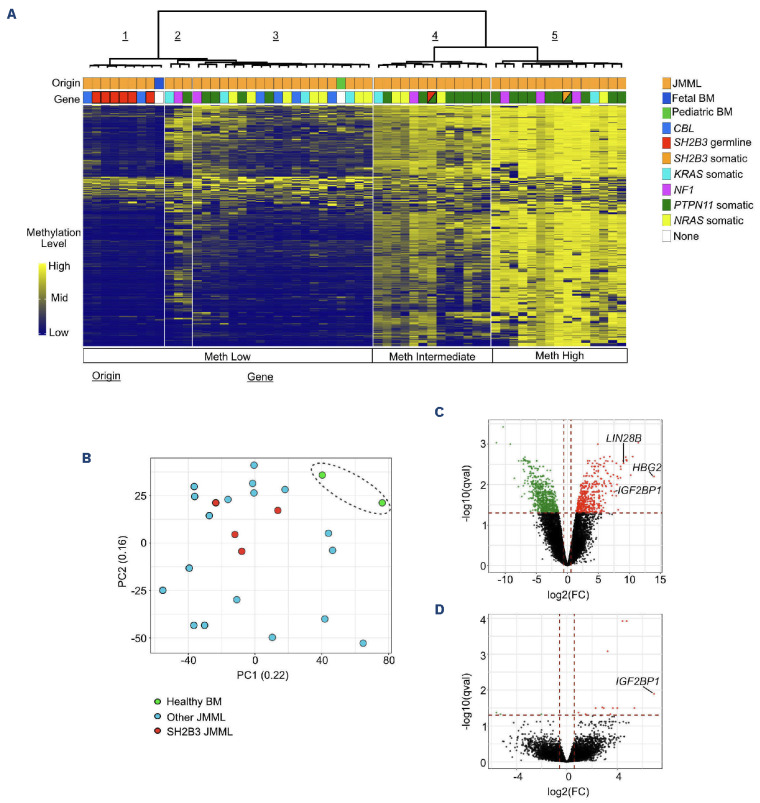
Comment in
-
SH2B3 alterations in a novel genetic condition, juvenile myelomonocytic leukemia, and myeloproliferative neoplasia.Haematologica. 2024 Aug 1;109(8):2391-2394. doi: 10.3324/haematol.2023.284747. Haematologica. 2024. PMID: 38618667 Free PMC article. No abstract available.
References
-
- Chang TY, Dvorak CC, Loh ML. Bedside to bench in juvenile myelomonocytic leukemia: insights into leukemogenesis from a rare pediatric leukemia. Blood. 2014;124(16):2487-2497. - PubMed
-
- Locatelli F, Niemeyer CM. How I treat juvenile myelomonocytic leukemia. Blood. 2015;125(7):1083-1090. - PubMed
-
- Locatelli F, Nöllke P, Zecca M, et al. Hematopoietic stem cell transplantation (HSCT) in children with juvenile myelomonocytic leukemia (JMML): results of the EWOG-MDS/ EBMT trial. Blood. 2005;105(1):410-419. - PubMed
-
- Lapidot T, Grunberger T, Vormoor J, et al. Identification of human juvenile chronic myelogenous leukemia stem cells capable of initiating the disease in primary and secondary SCID mice. Blood. 1996;88(7):2655-2664. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
