

A PERK-Specific Inhibitor Blocks Metastatic Progression by Limiting Integrated Stress Response-Dependent Survival of Quiescent Cancer Cells
- PMID: 37982738
- PMCID: PMC10842363
- DOI: 10.1158/1078-0432.CCR-23-1427
A PERK-Specific Inhibitor Blocks Metastatic Progression by Limiting Integrated Stress Response-Dependent Survival of Quiescent Cancer Cells
Abstract
Purpose: The integrated stress response (ISR) kinase PERK serves as a survival factor for both proliferative and dormant cancer cells. We aim to validate PERK inhibition as a new strategy to specifically eliminate solitary disseminated cancer cells (DCC) in secondary sites that eventually reawake and originate metastasis.
Experimental design: A novel clinical-grade PERK inhibitor (HC4) was tested in mouse syngeneic and PDX models that present quiescent/dormant DCCs or growth-arrested cancer cells in micro-metastatic lesions that upregulate ISR.
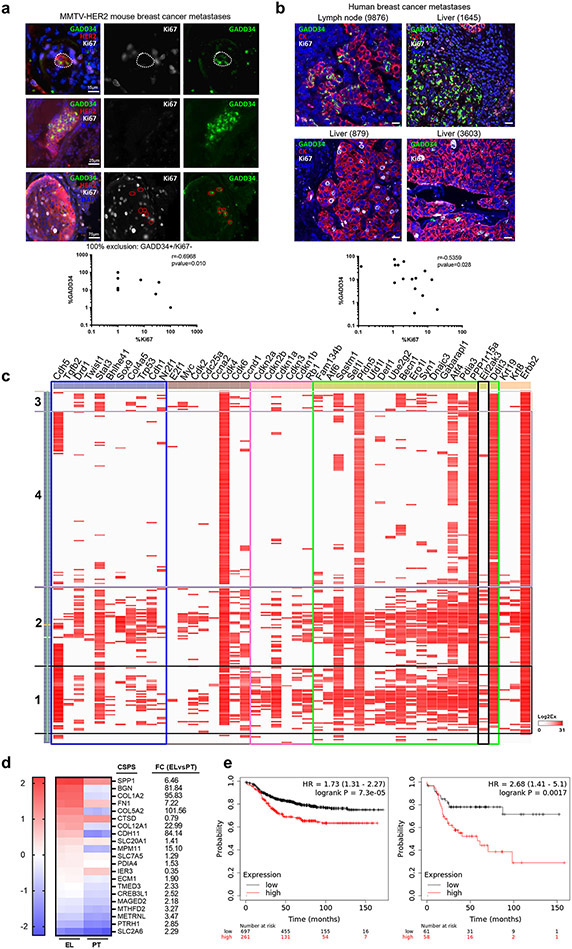
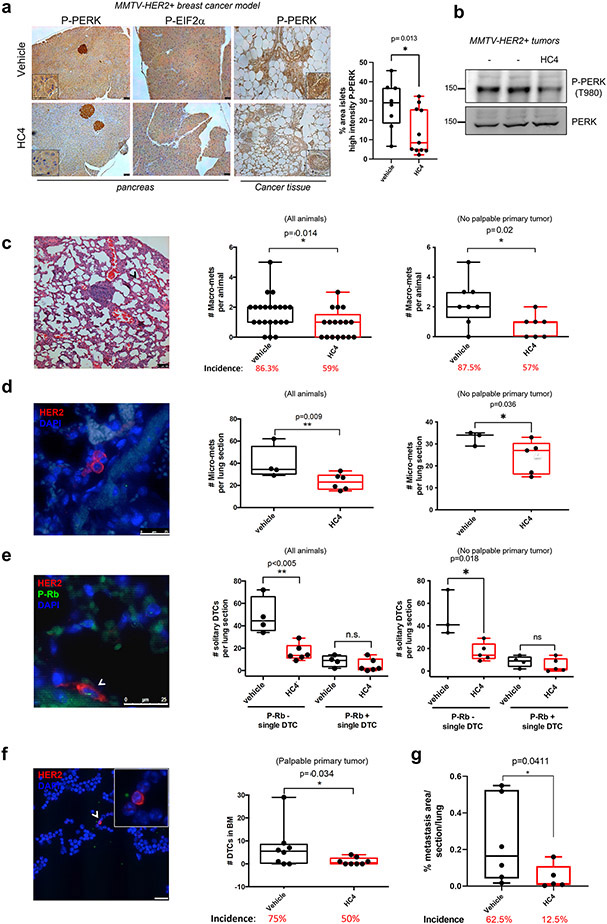
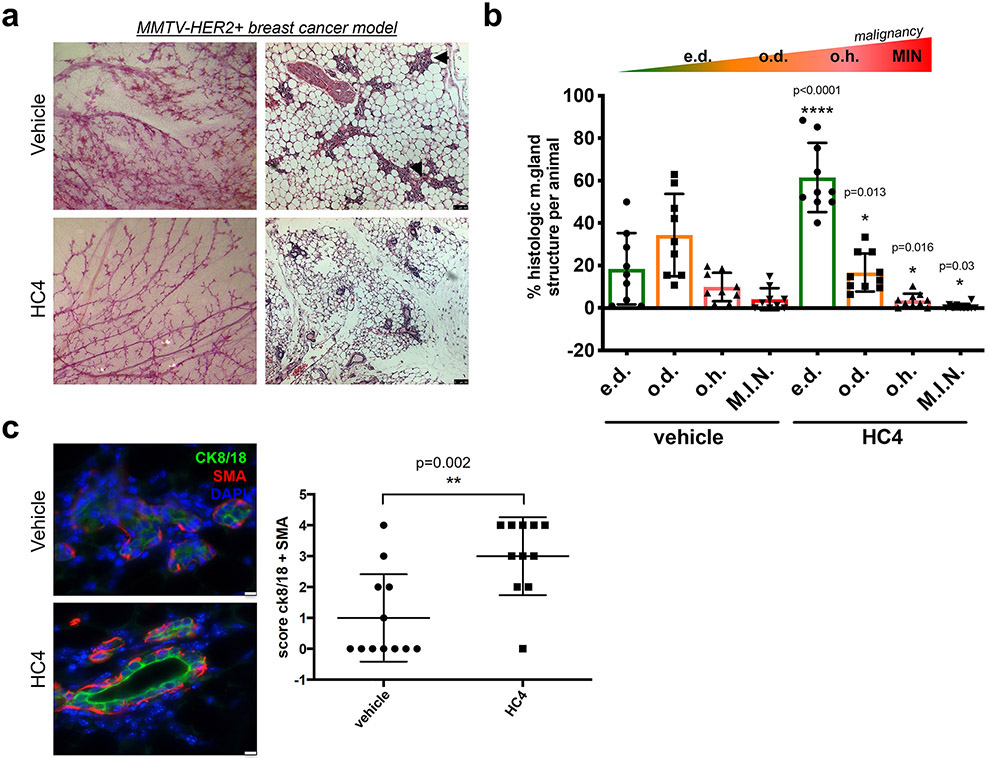
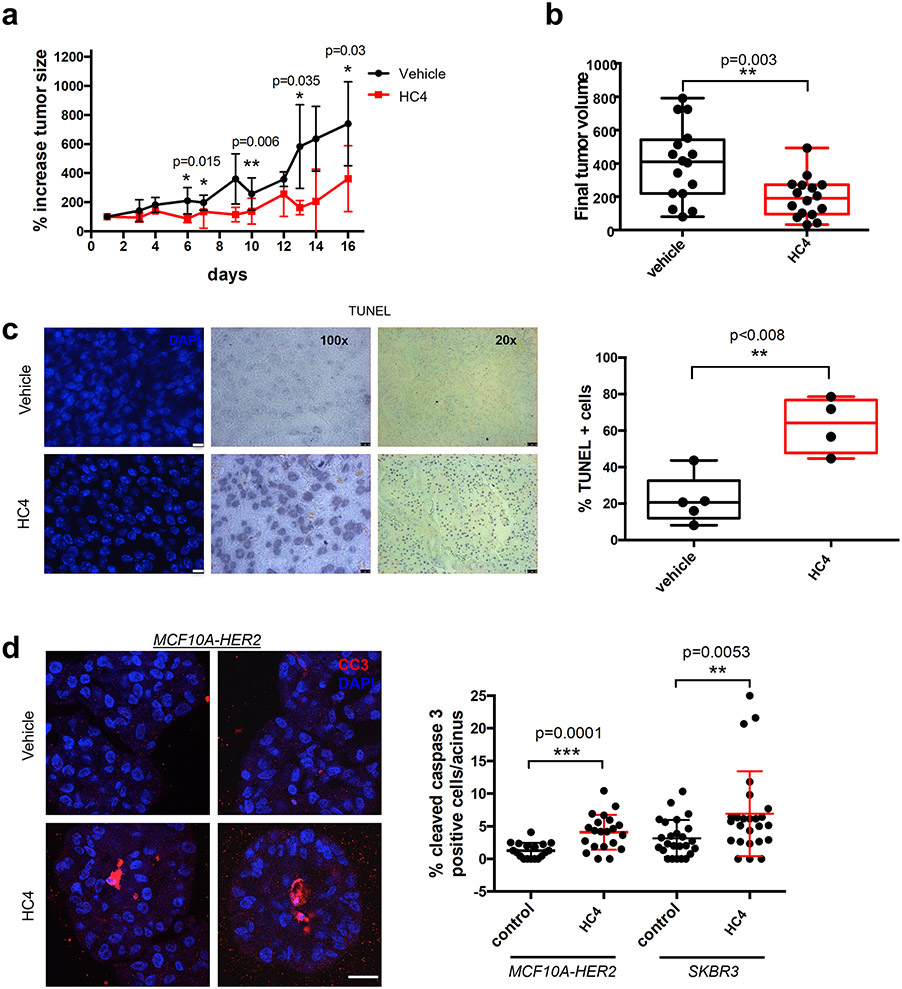
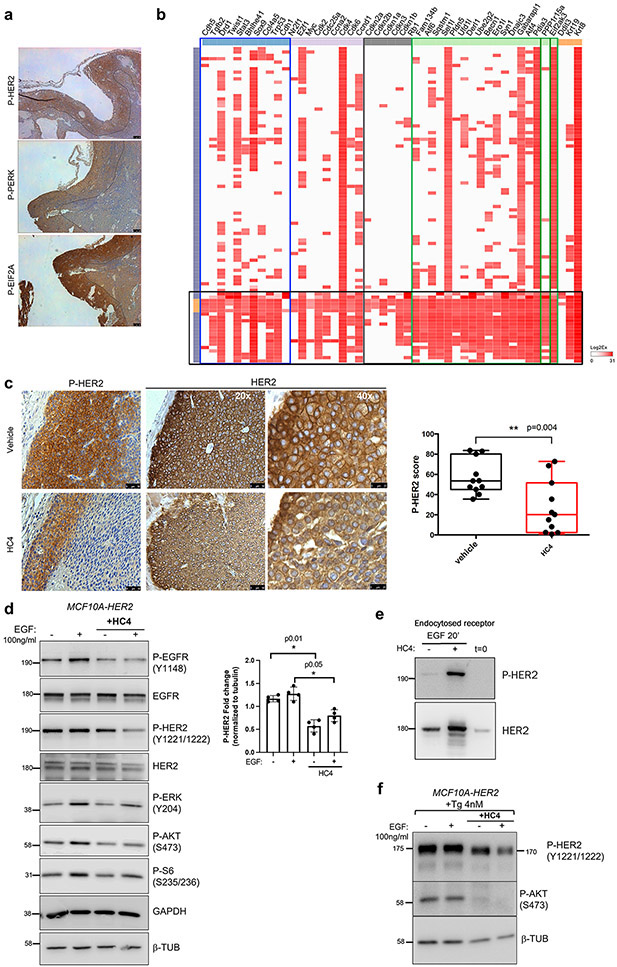
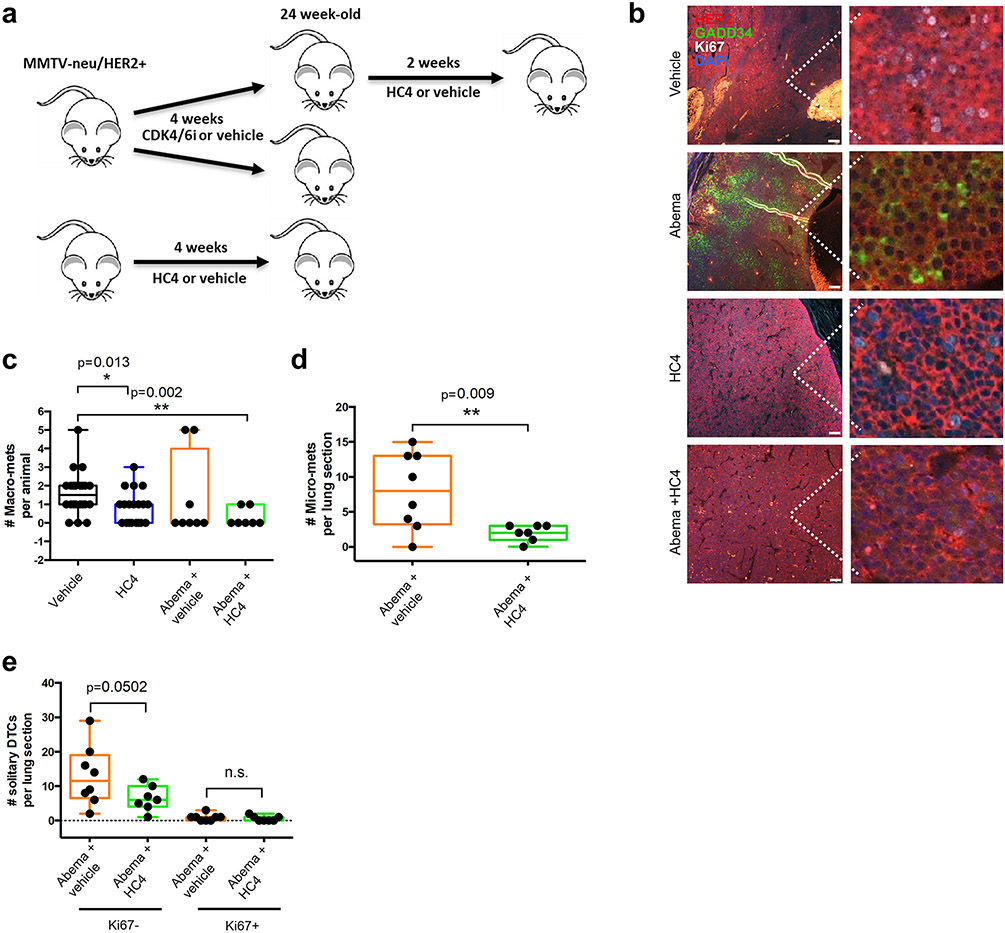
Results: HC4 significantly blocks metastasis, by killing quiescent/slow-cycling ISRhigh, but not proliferative ISRlow DCCs. HC4 blocked expansion of established micro-metastasis that contained ISRhigh slow-cycling cells. Single-cell gene expression profiling and imaging revealed that a significant proportion of solitary DCCs in lungs were indeed dormant and displayed an unresolved ER stress as revealed by high expression of a PERK-regulated signature. In human breast cancer metastasis biopsies, GADD34 expression (PERK-regulated gene) and quiescence were positively correlated. HC4 effectively eradicated dormant bone marrow DCCs, which usually persist after rounds of therapies. Importantly, treatment with CDK4/6 inhibitors (to force a quiescent state) followed by HC4 further reduced metastatic burden. In HNSCC and HER2+ cancers HC4 caused cell death in dormant DCCs. In HER2+ tumors, PERK inhibition caused killing by reducing HER2 activity because of sub-optimal HER2 trafficking and phosphorylation in response to EGF.
Conclusions: Our data identify PERK as a unique vulnerability in quiescent or slow-cycling ISRhigh DCCs. The use of PERK inhibitors may allow targeting of pre-existing or therapy-induced growth arrested "persister" cells that escape anti-proliferative therapies.
©2023 American Association for Cancer Research.
Conflict of interest statement
Conflict of Interests
JAAG is a scientific co-founder of, scientific advisory board member and equity owner in HiberCell and receives financial compensation as a consultant for HiberCell, a Mount Sinai spin-off company focused on therapeutics that prevent or delay cancer recurrence. VC, EFF, SF, DL, AN, MES and MM are HiberCell employees. The remaining authors declare no conflicts of interest.
Figures
References
-
- Chevet E, Hetz C, Samali A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov 2015;5(6):586–97 doi 10.1158/2159-8290.CD-14-1490. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
