

This is a preprint.
Transcriptome-wide analysis of the function of Ded1 in translation preinitiation complex assembly in a reconstituted in vitro system
- PMID: 37986768
- PMCID: PMC10659408
- DOI: 10.1101/2023.10.16.562452
Transcriptome-wide analysis of the function of Ded1 in translation preinitiation complex assembly in a reconstituted in vitro system
Update in
-
Transcriptome-wide analysis of the function of Ded1 in translation preinitiation complex assembly in a reconstituted in vitro system.Elife. 2024 Apr 4;13:RP93255. doi: 10.7554/eLife.93255. Elife. 2024. PMID: 38573742 Free PMC article.
Abstract
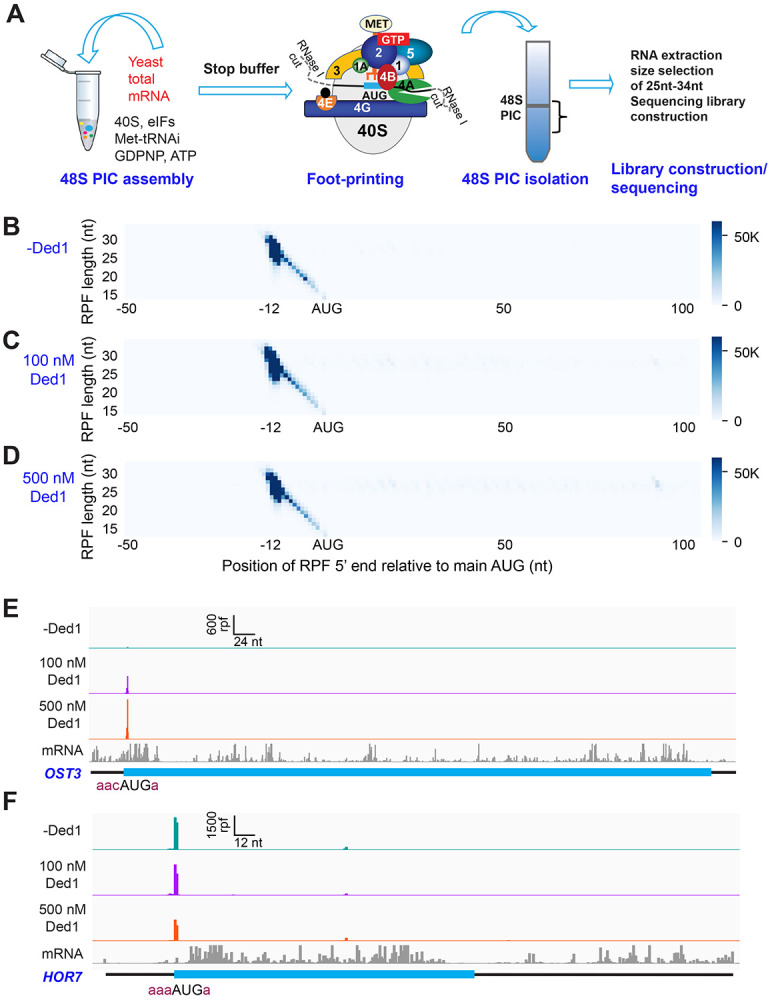
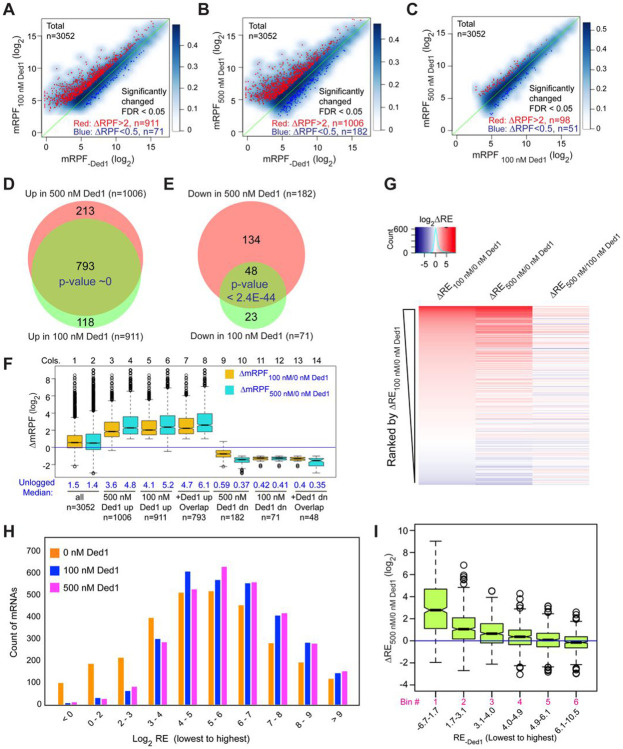
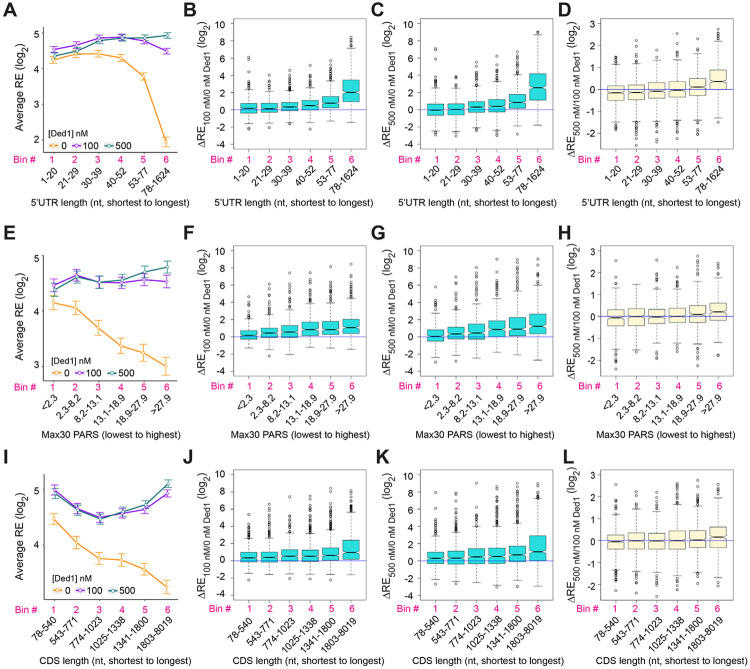
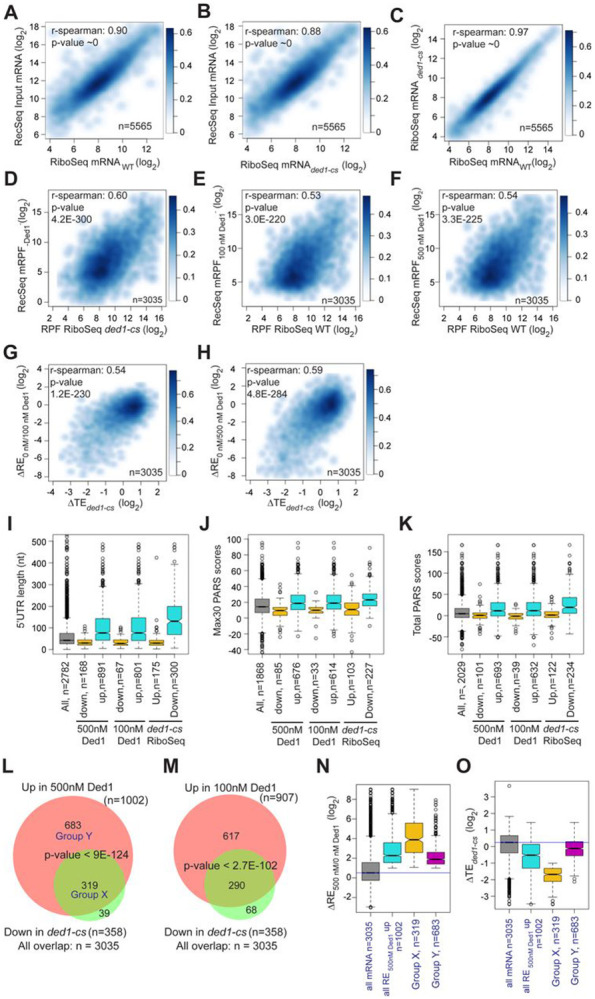
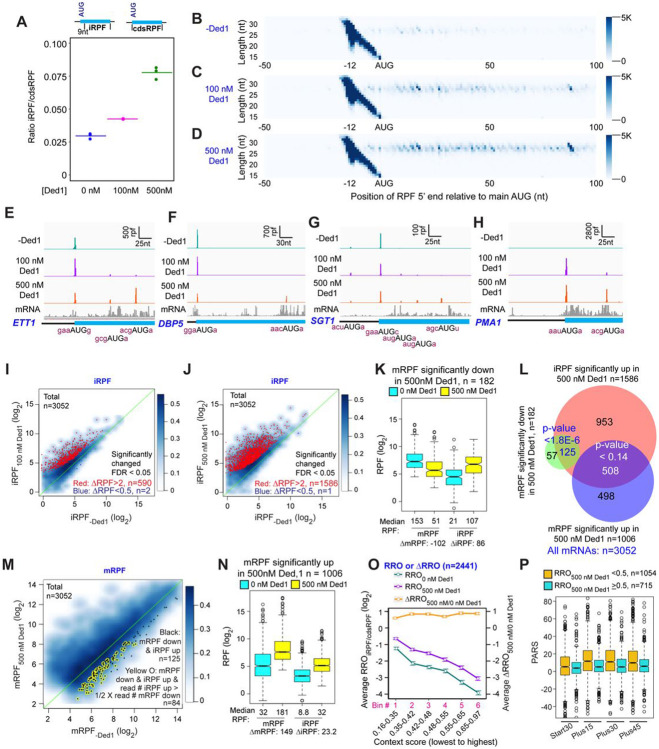
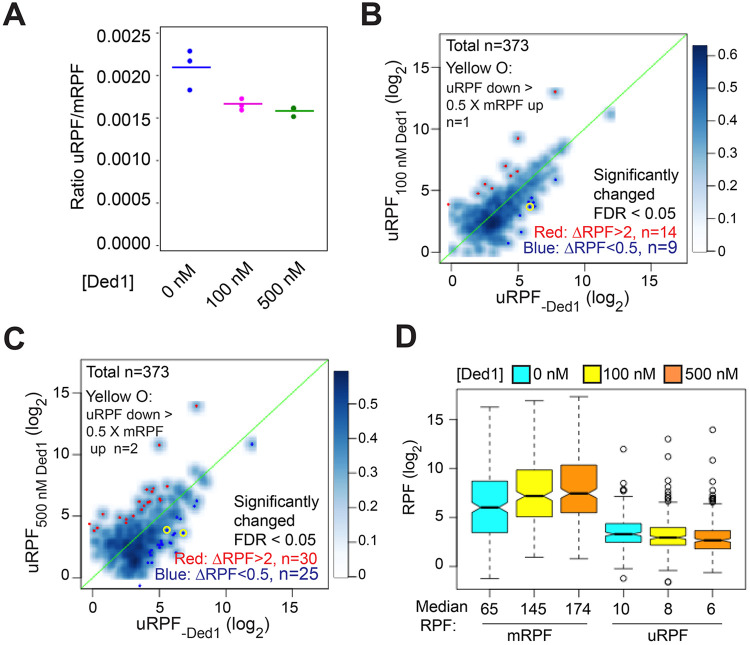
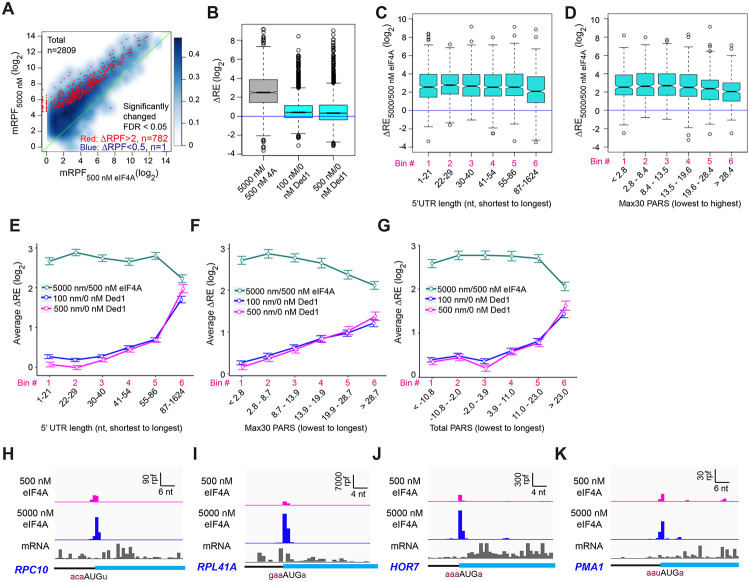
We have developed a deep sequencing-based approach, Rec-Seq, that allows simultaneous monitoring of ribosomal 48S pre-initiation complex (PIC) formation on every mRNA in the translatome in an in vitro reconstituted system. Rec-Seq isolates key early steps in translation initiation in the absence of all other cellular components and processes. Using this approach we show that the DEAD-box ATPase Ded1 promotes 48S PIC formation on the start codons of >1000 native mRNAs, most of which have long, structured 5'-untranslated regions (5'UTRs). Remarkably, initiation measured in Rec-Seq was enhanced by Ded1 for most mRNAs previously shown to be highly Ded1-dependent by ribosome profiling of ded1 mutants in vivo, demonstrating that the core translation functions of the factor are recapitulated in the purified system. Our data do not support a model in which Ded1acts by reducing initiation at alternative start codons in 5'UTRs and instead indicate it functions by directly promoting mRNA recruitment to the 43S PIC and scanning to locate the main start codon. We also provide evidence that eIF4A, another essential DEAD-box initiation factor, is required for efficient PIC assembly on almost all mRNAs, regardless of their structural complexity, in contrast to the preferential stimulation by Ded1 of initiation on mRNAs with long, structured 5'UTRs.
Figures
Similar articles
-
Transcriptome-wide analysis of the function of Ded1 in translation preinitiation complex assembly in a reconstituted in vitro system.Elife. 2024 Apr 4;13:RP93255. doi: 10.7554/eLife.93255. Elife. 2024. PMID: 38573742 Free PMC article.
-
Yeast Ded1 promotes 48S translation pre-initiation complex assembly in an mRNA-specific and eIF4F-dependent manner.Elife. 2018 Oct 3;7:e38892. doi: 10.7554/eLife.38892. Elife. 2018. PMID: 30281017 Free PMC article.
-
Functional interplay between DEAD-box RNA helicases Ded1 and Dbp1 in preinitiation complex attachment and scanning on structured mRNAs in vivo.Nucleic Acids Res. 2019 Sep 19;47(16):8785-8806. doi: 10.1093/nar/gkz595. Nucleic Acids Res. 2019. PMID: 31299079 Free PMC article.
-
Structural Insights into the Mechanism of Scanning and Start Codon Recognition in Eukaryotic Translation Initiation.Trends Biochem Sci. 2017 Aug;42(8):589-611. doi: 10.1016/j.tibs.2017.03.004. Epub 2017 Apr 22. Trends Biochem Sci. 2017. PMID: 28442192 Review.
-
Cap-dependent, scanning-free translation initiation mechanisms.Biochim Biophys Acta. 2015 Nov;1849(11):1313-8. doi: 10.1016/j.bbagrm.2015.09.006. Epub 2015 Sep 14. Biochim Biophys Acta. 2015. PMID: 26381322 Review.
References
-
- Acker MG, Kolitz SE, Mitchell SF, Nanda JS, Lorsch JR. 2007. Reconstitution of yeast translation initiation. Methods Enzymol 430: 111–145. - PubMed
-
- Archer SK, Shirokikh NE, Beilharz TH, Preiss T. 2016. Dynamics of ribosome scanning and recycling revealed by translation complex profiling. Nature 535: 570–574. - PubMed
-
- Berthelot K, Muldoon M, Rajkowitsch L, Hughes J, McCarthy JE. 2004. Dynamics and processivity of 40S ribosome scanning on mRNA in yeast. Mol Microbiol 51: 987–1001. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous