

This is a preprint.
Improving Hi-C contact matrices using genome graphs
- PMID: 37986943
- PMCID: PMC10659349
- DOI: 10.1101/2023.11.08.566275
Improving Hi-C contact matrices using genome graphs
Abstract
Three-dimensional chromosome structure plays an important role in fundamental genomic functions. Hi-C, a high-throughput, sequencing-based technique, has drastically expanded our comprehension of 3D chromosome structures. The first step of Hi-C analysis pipeline involves mapping sequencing reads from Hi-C to linear reference genomes. However, the linear reference genome does not incorporate genetic variation information, which can lead to incorrect read alignments, especially when analyzing samples with substantial genomic differences from the reference such as cancer samples. Using genome graphs as the reference facilitates more accurate mapping of reads, however, new algorithms are required for inferring linear genomes from Hi-C reads mapped on genome graphs and constructing corresponding Hi-C contact matrices, which is a prerequisite for the subsequent steps of the Hi-C analysis such as identifying topologically associated domains and calling chromatin loops. We introduce the problem of genome sequence inference from Hi-C data mediated by genome graphs. We formalize this problem, show the hardness of solving this problem, and introduce a novel heuristic algorithm specifically tailored to this problem. We provide a theoretical analysis to evaluate the efficacy of our algorithm. Finally, our empirical experiments indicate that the linear genomes inferred from our method lead to the creation of improved Hi-C contact matrices. These enhanced matrices show a reduction in erroneous patterns caused by structural variations and are more effective in accurately capturing the structures of topologically associated domains.
Figures
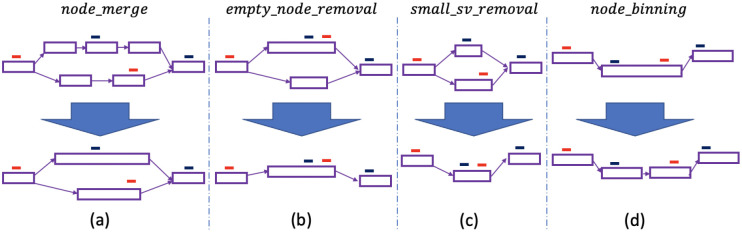

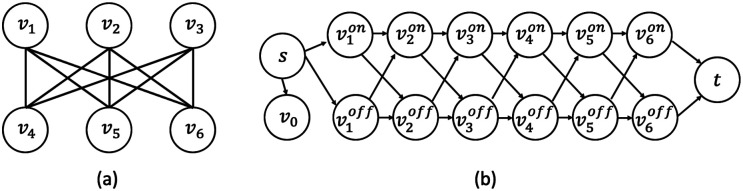


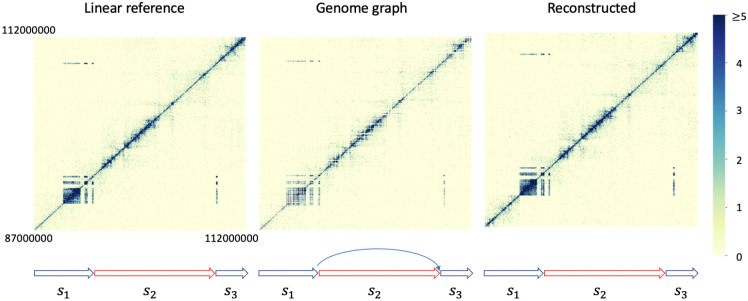



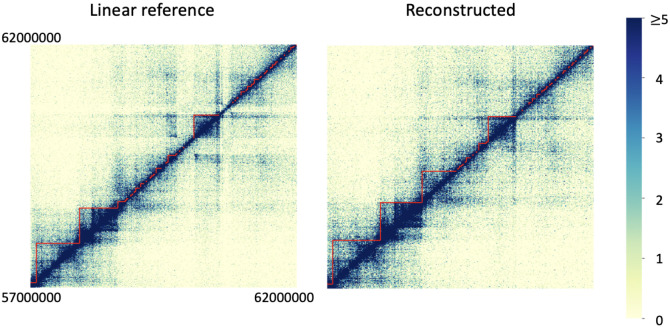

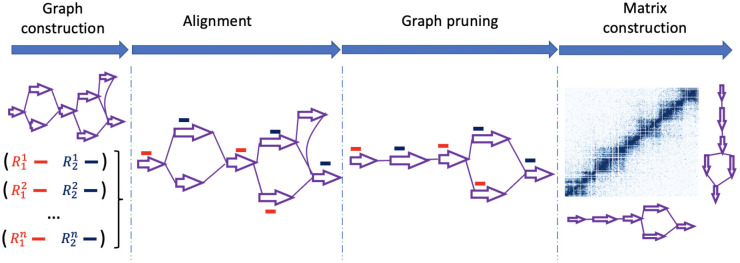
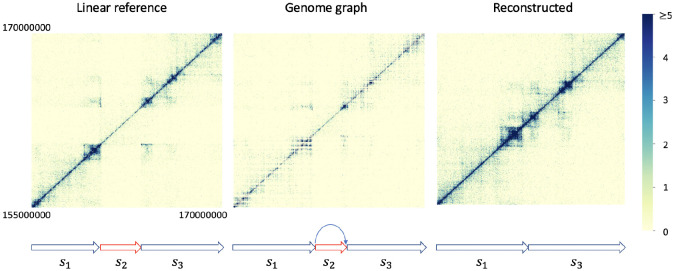
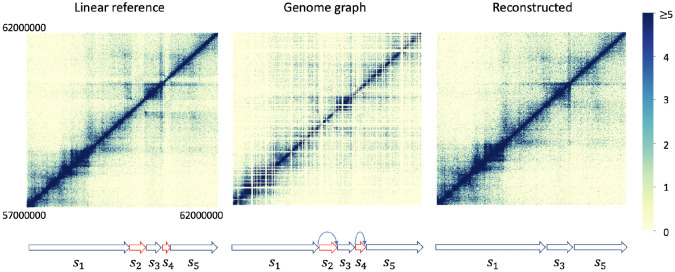

References
-
- Fraser Peter and Bickmore Wendy. Nuclear organization of the genome and the potential for gene regulation. Nature, 447(7143):413–417, 2007. - PubMed
-
- Grewal Shiv IS and Moazed Danesh. Heterochromatin and epigenetic control of gene expression. Science, 301(5634):798–802, 2003. - PubMed
-
- Lieberman-Aiden Erez, Van Berkum Nynke L, Williams Louise, Imakaev Maxim, Ragoczy Tobias, Telling Agnes, Amit Ido, Lajoie Bryan R, Sabo Peter J, Dorschner Michael O, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science, 326(5950):289–293, 2009. - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources