

This is a preprint.
Deciphering epistatic genetic regulation of cardiac hypertrophy
- PMID: 37987017
- PMCID: PMC10659487
- DOI: 10.1101/2023.11.06.23297858
Deciphering epistatic genetic regulation of cardiac hypertrophy
Update in
-
Epistasis regulates genetic control of cardiac hypertrophy.Nat Cardiovasc Res. 2025 Jun;4(6):740-760. doi: 10.1038/s44161-025-00656-8. Epub 2025 Jun 5. Nat Cardiovasc Res. 2025. PMID: 40473955 Free PMC article.
Abstract
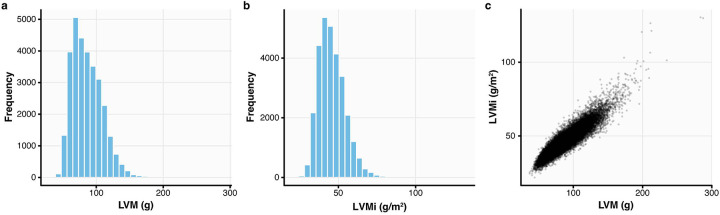
Although genetic variant effects often interact non-additively, strategies to uncover epistasis remain in their infancy. Here, we develop low-signal signed iterative random forests to elucidate the complex genetic architecture of cardiac hypertrophy, using deep learning-derived left ventricular mass estimates from 29,661 UK Biobank cardiac MRIs. We report epistatic variants near CCDC141, IGF1R, TTN, and TNKS, identifying loci deemed insignificant in genome-wide association studies. Functional genomic and integrative enrichment analyses reveal that genes mapped from these loci share biological process gene ontologies and myogenic regulatory factors. Transcriptomic network analyses using 313 human hearts demonstrate strong co-expression correlations among these genes in healthy hearts, with significantly reduced connectivity in failing hearts. To assess causality, RNA silencing in human induced pluripotent stem cell-derived cardiomyocytes, combined with novel microfluidic single-cell morphology analysis, confirms that cardiomyocyte hypertrophy is non-additively modifiable by interactions between CCDC141, TTN, and IGF1R. Our results expand the scope of cardiac genetic regulation to epistasis.
Conflict of interest statement
Competing interests E.A.A. is a founder of Personalis, Deepcell, Svexa, Candela, Saturnus Bio, and Parameter Health; an advisor to SequenceBio, Foresite Labs, Pacific Biosciences, and Versant Ventures; a non-executive director for AstraZeneca and Svexa; a stockholder in Pacific Biosciences and AstraZeneca; and has received in-kind collaborative support from Illumina, Pacific Biosciences, Oxford Nanopore, Cache, and Cellsonics. V.N.P. is an SAB member for and receives research support from BioMarin, Inc., an SAB member for Lexeo Therapeutics, and a consultant for Constantiam Biosciences and viz.ai. C.S.W. is a consultant for AiRNA Bio and Avidity Biosciences. While these companies may have interests in genomics or cardiovascular health, they had no direct role in the design, data presentation, analysis, or interpretation of this study. The remaining authors declare no competing interest.
Figures















References
-
- Burkhoff D., Mirsky I. & Suga H. Assessment of systolic and diastolic ventricular properties via pressure-volume analysis: a guide for clinical, translational, and basic researchers. American Journal of Physiology-Heart and Circulatory Physiology 289, H501–H512 (2005). - PubMed
-
- Haider A. W., Larson M. G., Benjamin E. J. & Levy D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J. Am. Coll. Cardiol. 32, 1454–1459 (1998). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources