

SETD2 deficiency accelerates sphingomyelin accumulation and promotes the development of renal cancer
- PMID: 37989747
- PMCID: PMC10663509
- DOI: 10.1038/s41467-023-43378-w
SETD2 deficiency accelerates sphingomyelin accumulation and promotes the development of renal cancer
Abstract
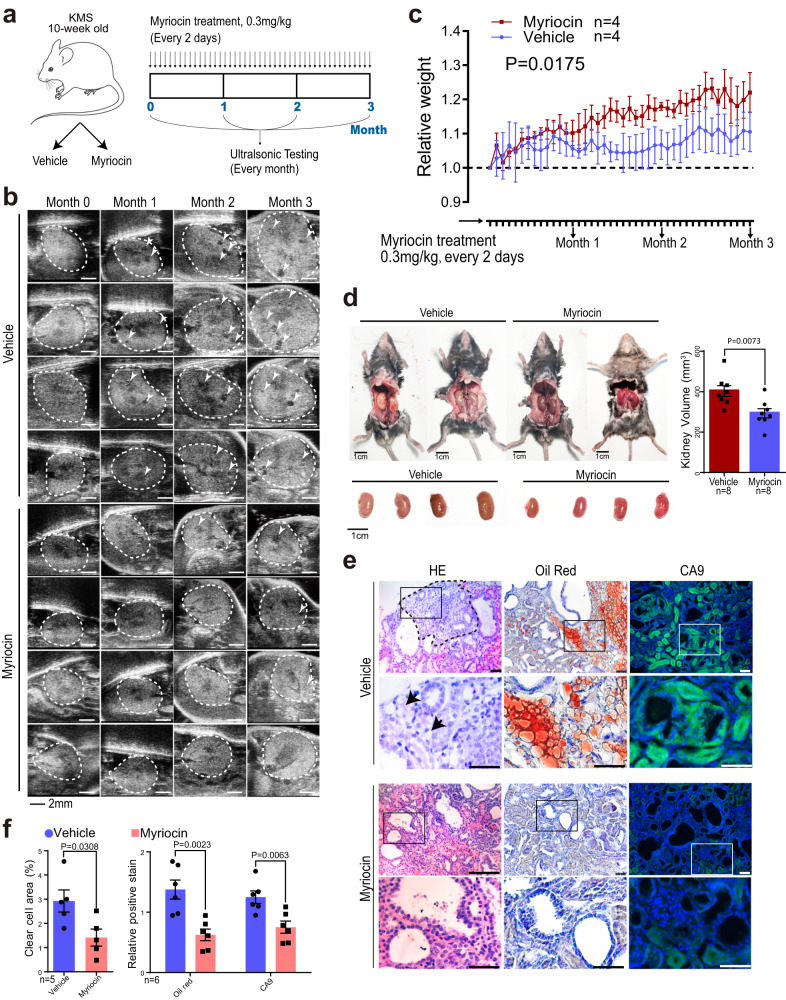
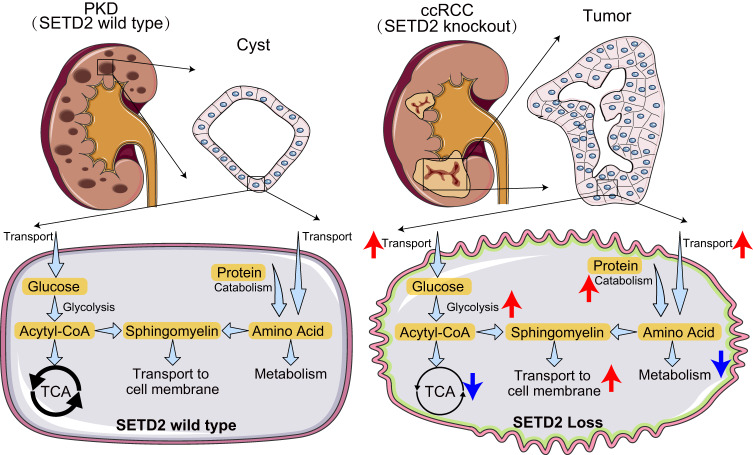
Patients with polycystic kidney disease (PKD) encounter a high risk of clear cell renal cell carcinoma (ccRCC), a malignant tumor with dysregulated lipid metabolism. SET domain-containing 2 (SETD2) has been identified as an important tumor suppressor and an immunosuppressor in ccRCC. However, the role of SETD2 in ccRCC generation in PKD remains largely unexplored. Herein, we perform metabolomics, lipidomics, transcriptomics and proteomics within SETD2 loss induced PKD-ccRCC transition mouse model. Our analyses show that SETD2 loss causes extensive metabolic reprogramming events that eventually results in enhanced sphingomyelin biosynthesis and tumorigenesis. Clinical ccRCC patient specimens further confirm the abnormal metabolic reprogramming and sphingomyelin accumulation. Tumor symptom caused by Setd2 knockout is relieved by myriocin, a selective inhibitor of serine-palmitoyl-transferase and sphingomyelin biosynthesis. Our results reveal that SETD2 deficiency promotes large-scale metabolic reprogramming and sphingomyelin biosynthesis during PKD-ccRCC transition. This study introduces high-quality multi-omics resources and uncovers a regulatory mechanism of SETD2 on lipid metabolism during tumorigenesis.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





References
Publication types
MeSH terms
Substances
Grants and funding
- T2122002/National Natural Science Foundation of China (National Science Foundation of China)
- 22077079/National Natural Science Foundation of China (National Science Foundation of China)
- 82203255/National Natural Science Foundation of China (National Science Foundation of China)
- 82372604/National Natural Science Foundation of China (National Science Foundation of China)
- 82073104/National Natural Science Foundation of China (National Science Foundation of China)
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
