

Obesity, diabetes mellitus, and cardiometabolic risk: An Obesity Medicine Association (OMA) Clinical Practice Statement (CPS) 2023
- PMID: 37990743
- PMCID: PMC10661981
- DOI: 10.1016/j.obpill.2023.100056
Obesity, diabetes mellitus, and cardiometabolic risk: An Obesity Medicine Association (OMA) Clinical Practice Statement (CPS) 2023
Abstract
Background: This Obesity Medicine Association (OMA) Clinical Practice Statement (CPS) is intended to provide clinicians an overview of type 2 diabetes mellitus (T2DM), an obesity-related cardiometabolic risk factor.
Methods: The scientific support for this CPS is based upon published citations and clinical perspectives of OMA authors.
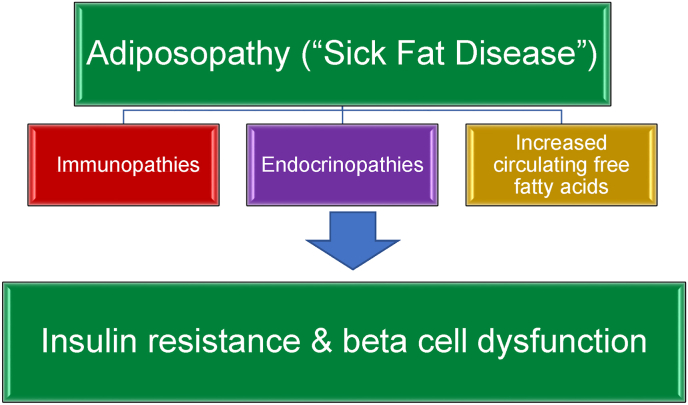
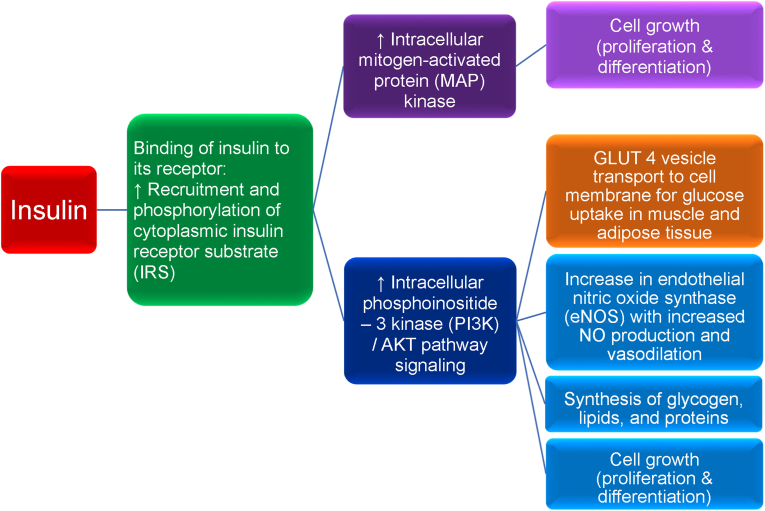
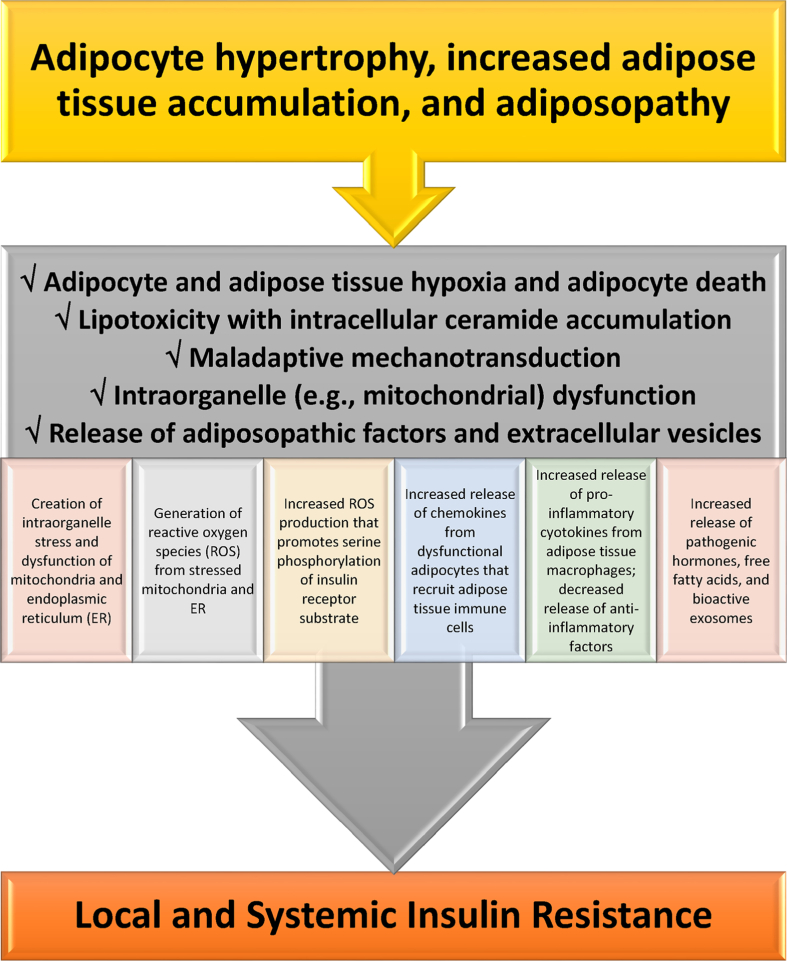
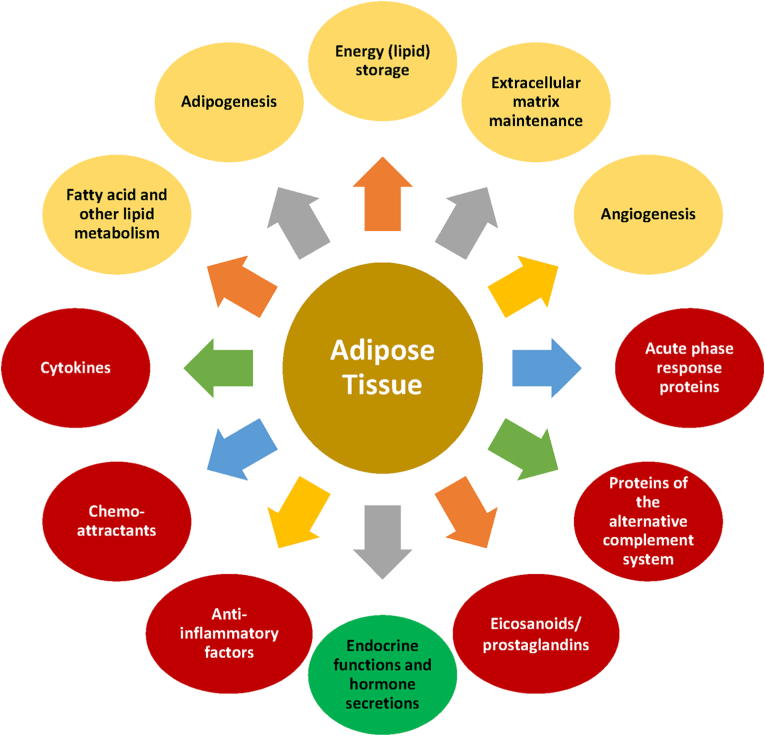
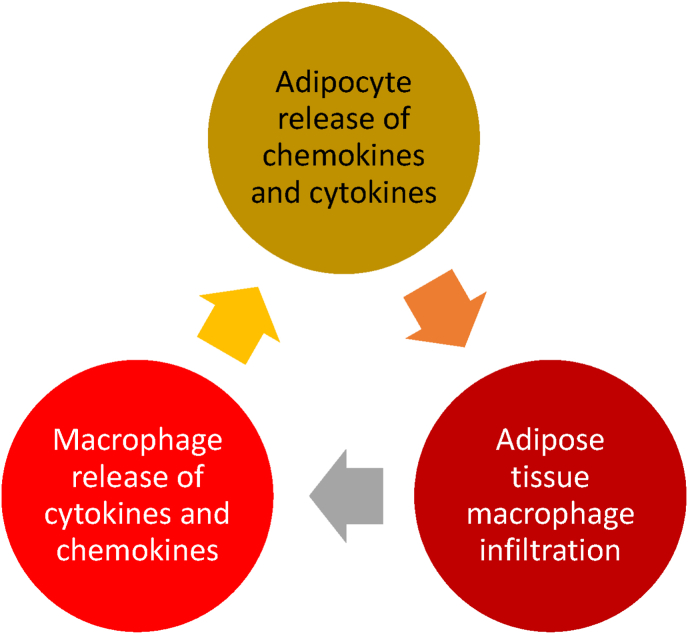
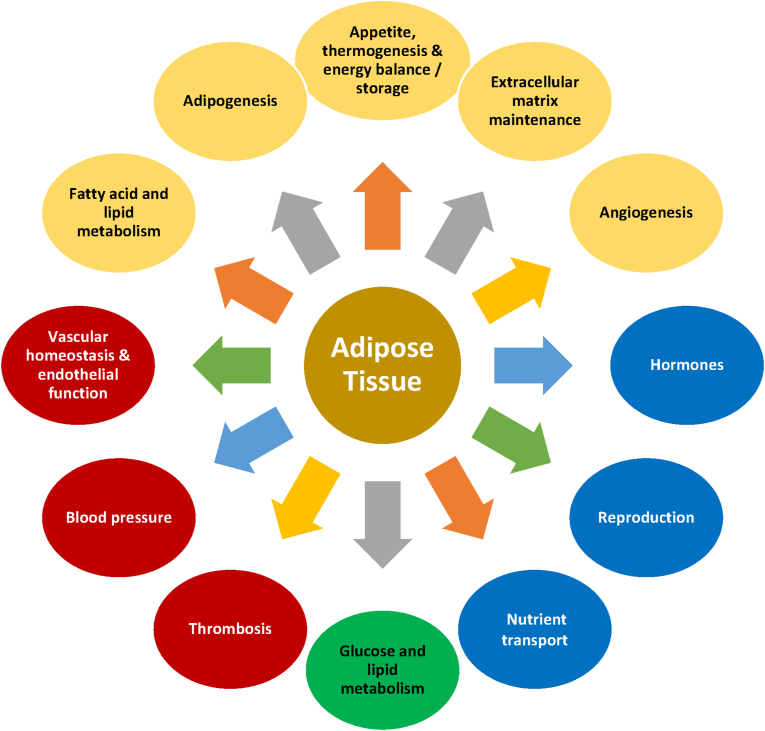
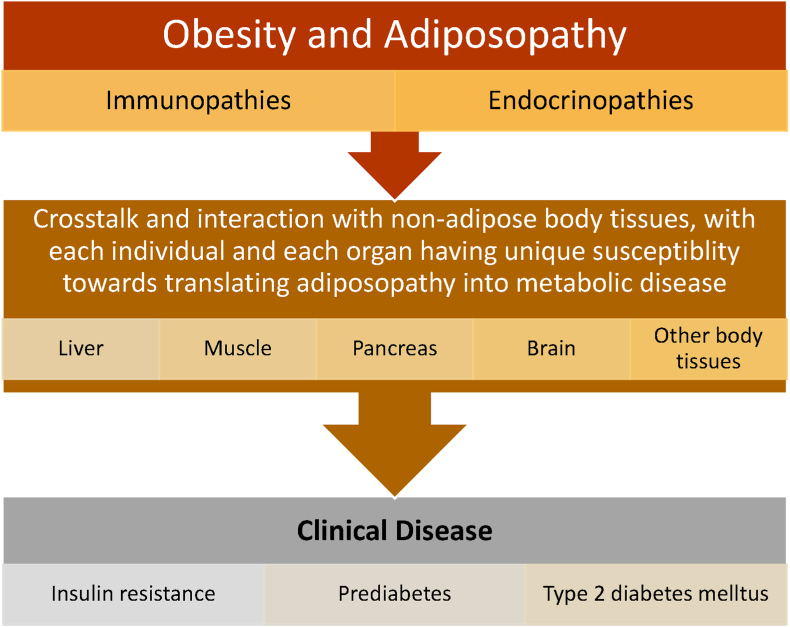
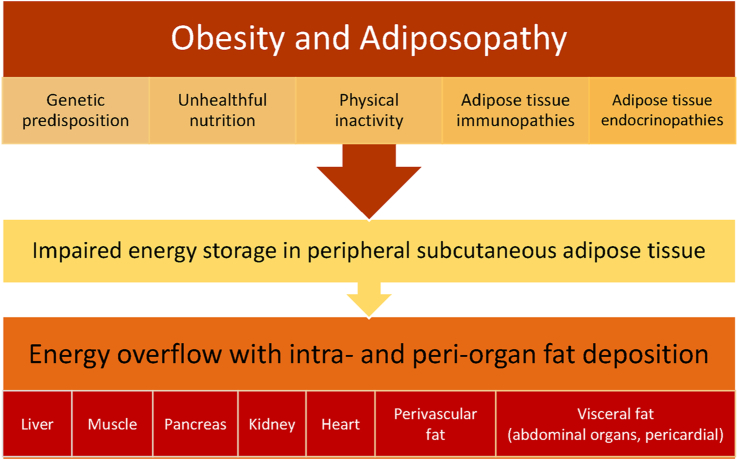
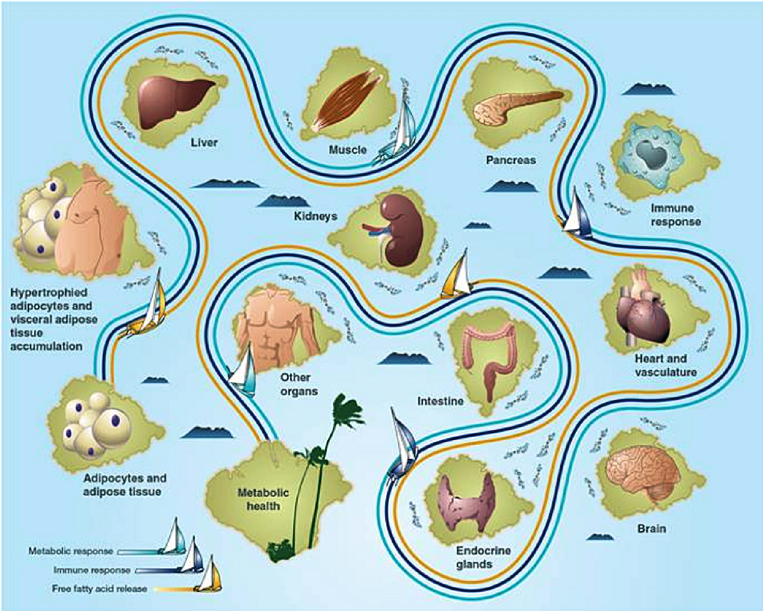
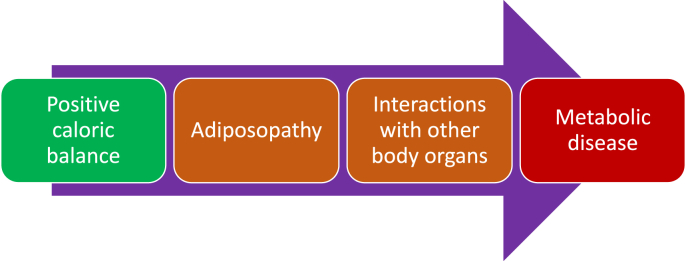
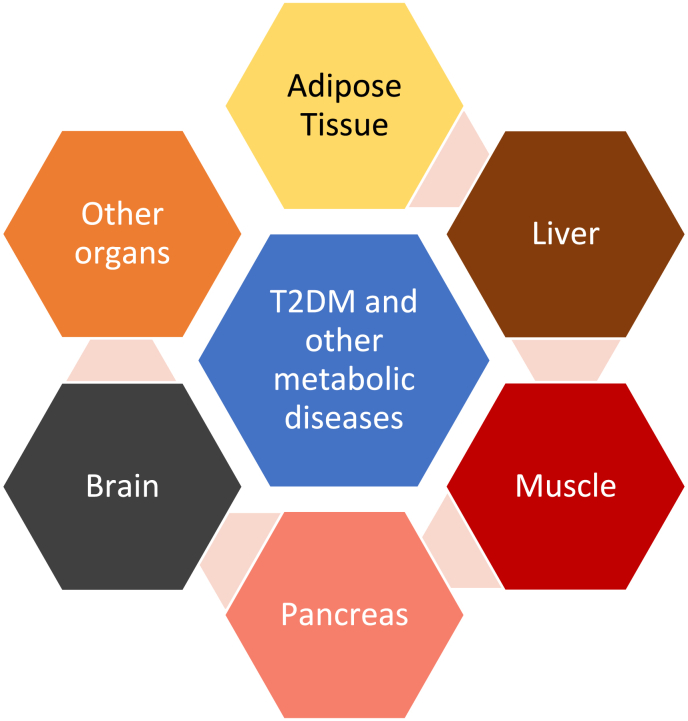
Results: Topics include T2DM and obesity as cardiometabolic risk factors, definitions of obesity and adiposopathy, and mechanisms for how obesity causes insulin resistance and beta cell dysfunction. Adipose tissue is an active immune and endocrine organ, whose adiposopathic obesity-mediated dysfunction contributes to metabolic abnormalities often encountered in clinical practice, including hyperglycemia (e.g., pre-diabetes mellitus and T2DM). The determination as to whether adiposopathy ultimately leads to clinical metabolic disease depends on crosstalk interactions and biometabolic responses of non-adipose tissue organs such as liver, muscle, pancreas, kidney, and brain.
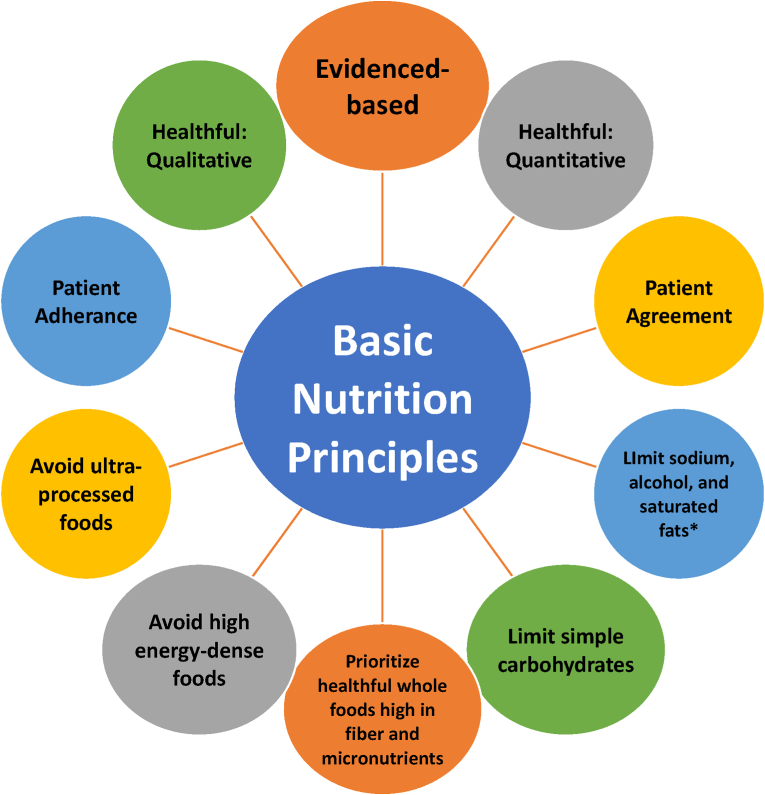
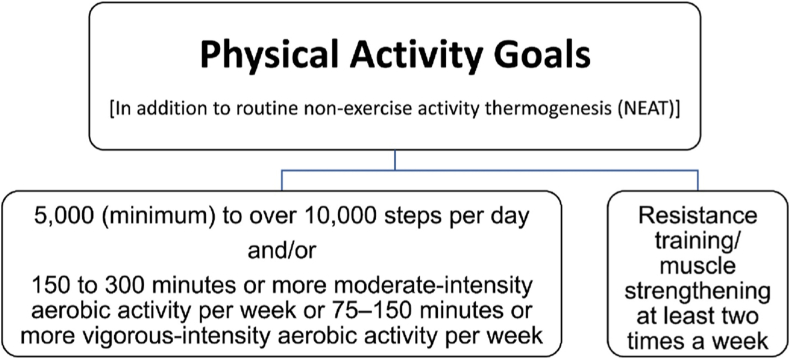
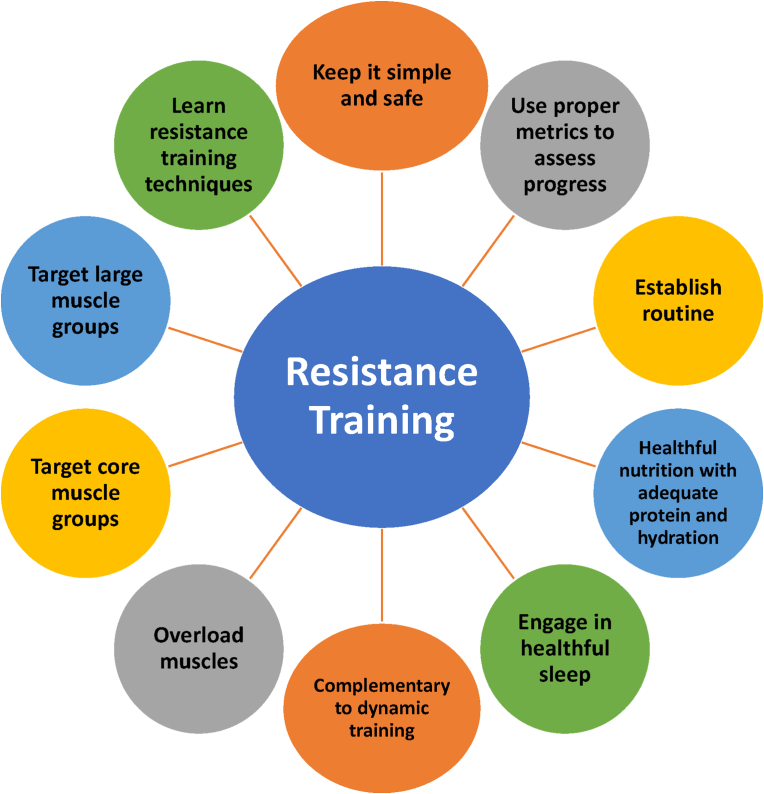
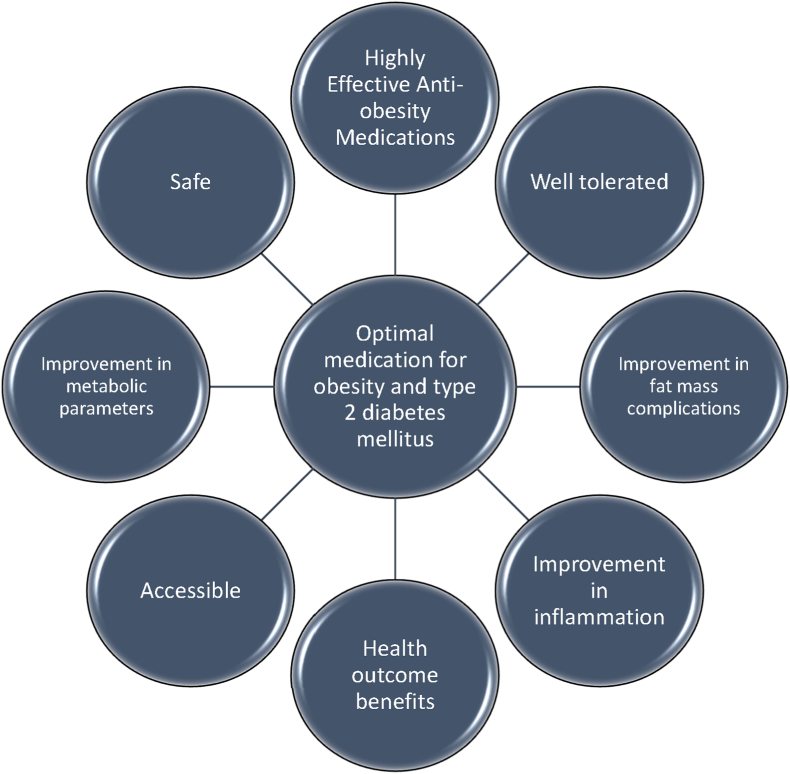
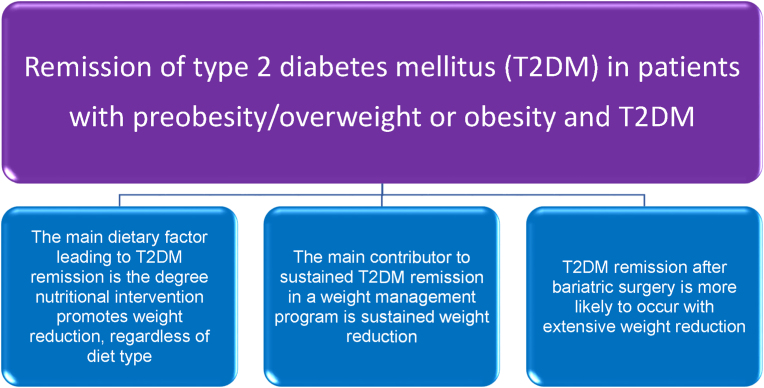
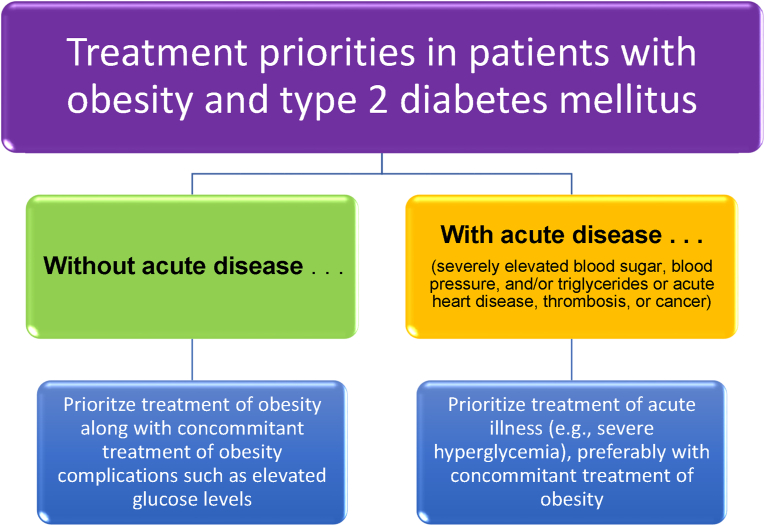
Conclusions: This review is intended to assist clinicians in the care of patients with the disease of obesity and T2DM. This CPS provides a simplified overview of how obesity may cause insulin resistance, pre-diabetes, and T2DM. It also provides an algorithmic approach towards treatment of a patient with obesity and T2DM, with "treat obesity first" as a priority. Finally, treatment of obesity and T2DM might best focus upon therapies that not only improve the weight of patients, but also improve the health outcomes of patients (e.g., cardiovascular disease and cancer).
Keywords: Adiposopathy; Cardiometabolic; Diabetes mellitus; Obesity.
© 2023 The Authors.
Figures


References
-
- Bays H.E., McCarthy W., Burridge K., Tondt J., Karjoo S., Christensen S., Ng J., Golden A., Davisson L., Richardson L. Obesity Algorithm eBook. www.obesityalgorithm.org.2021https://obesitymedicine.org/obesity-algorithm/ presented by the Obesity Medicine Association.
-
- Bays H., Abate N., Chandalia M. Adiposopathy: sick fat causes high blood sugar, high blood pressure and dyslipidemia. Future Cardiol. 2005;1:39–59. - PubMed
LinkOut - more resources
Full Text Sources
