

Development of Resistance to Type II JAK2 Inhibitors in MPN Depends on AXL Kinase and Is Targetable
- PMID: 37992313
- PMCID: PMC10831334
- DOI: 10.1158/1078-0432.CCR-23-0163
Development of Resistance to Type II JAK2 Inhibitors in MPN Depends on AXL Kinase and Is Targetable
Abstract
Purpose: Myeloproliferative neoplasms (MPN) dysregulate JAK2 signaling. Because clinical JAK2 inhibitors have limited disease-modifying effects, type II JAK2 inhibitors such as CHZ868 stabilizing inactive JAK2 and reducing MPN clones, gain interest. We studied whether MPN cells escape from type ll inhibition.
Experimental design: MPN cells were continuously exposed to CHZ868. We used phosphoproteomic analyses and ATAC/RNA sequencing to characterize acquired resistance to type II JAK2 inhibition, and targeted candidate mediators in MPN cells and mice.
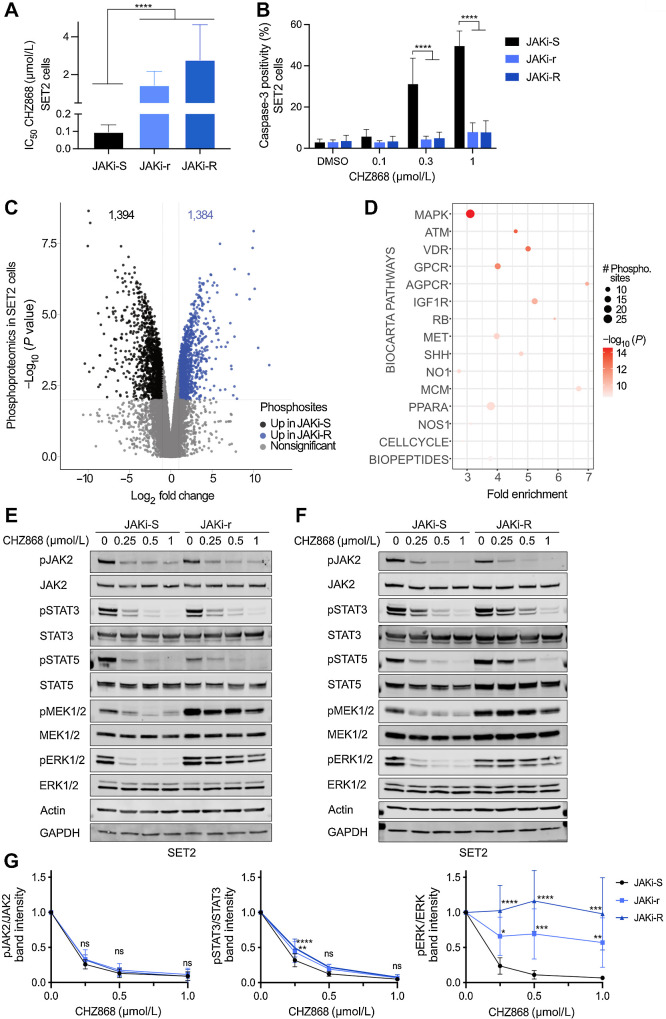
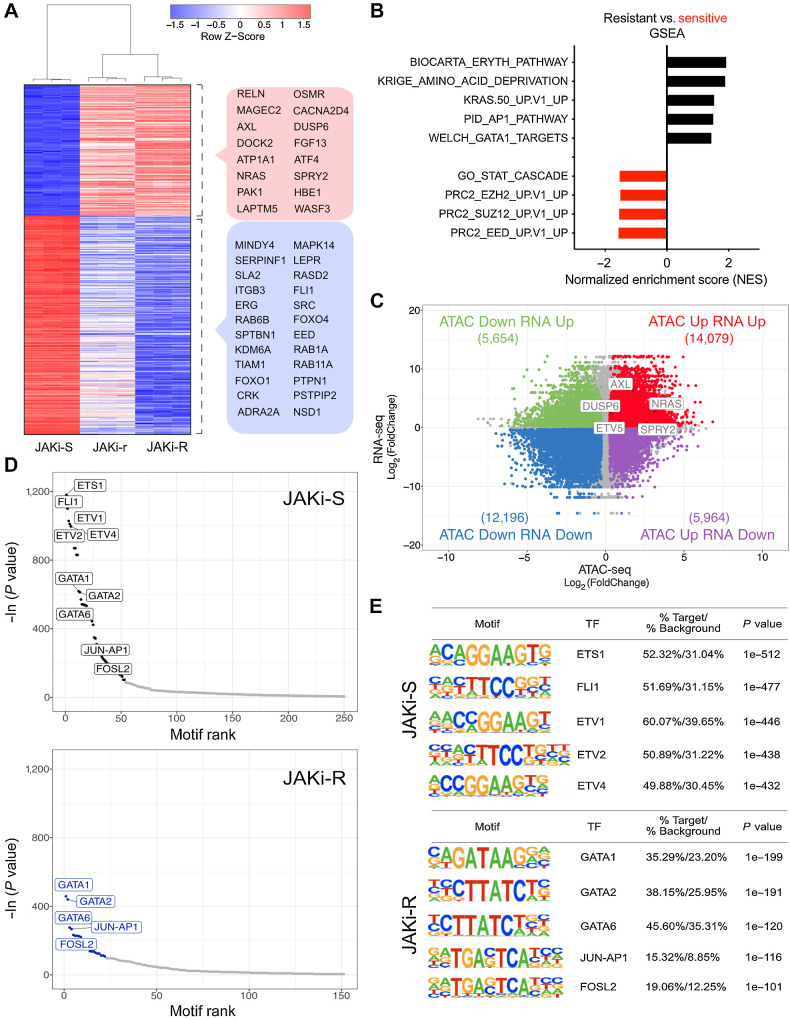
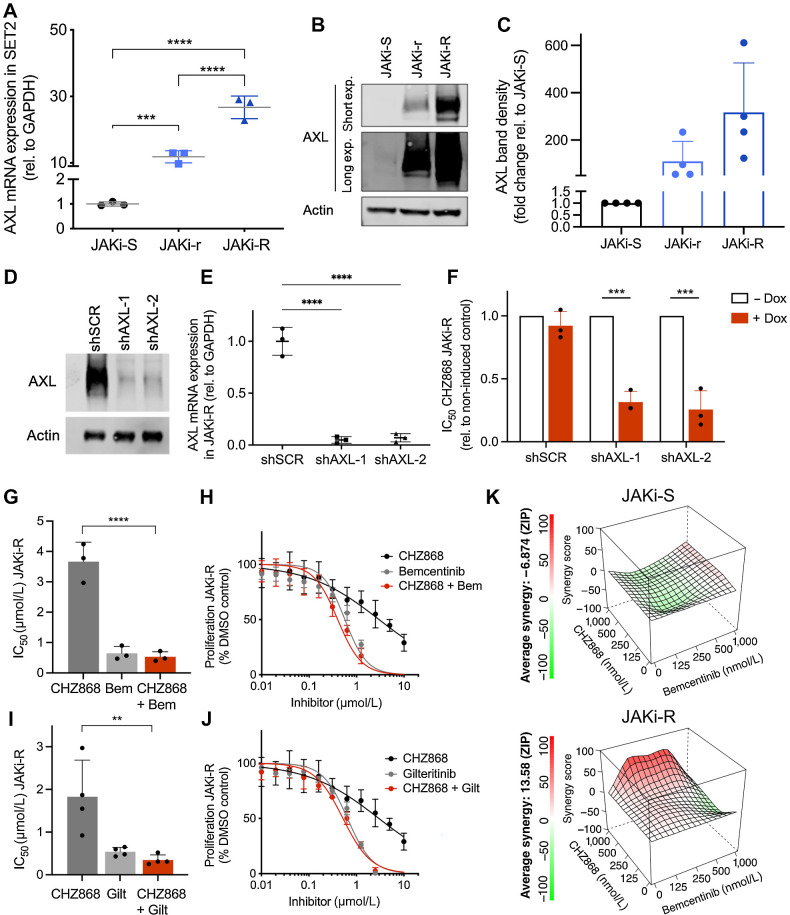
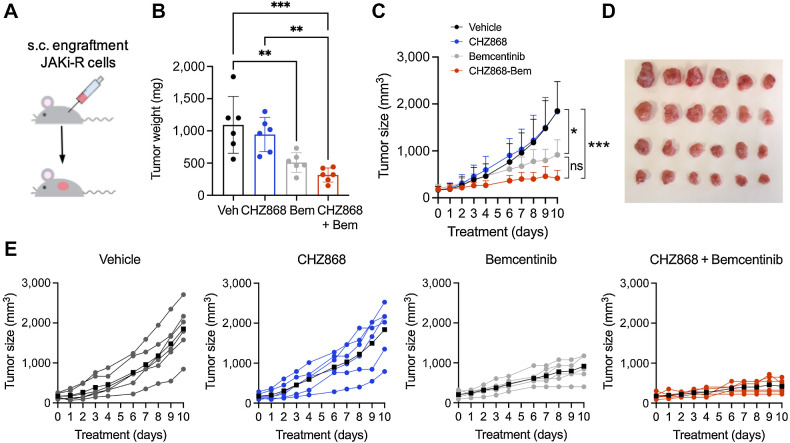
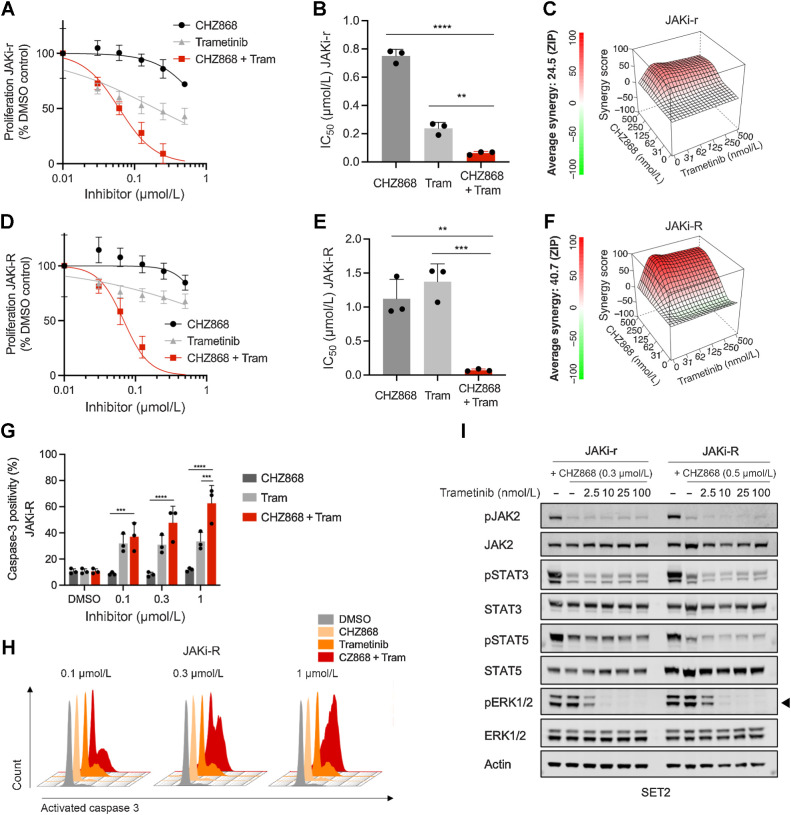
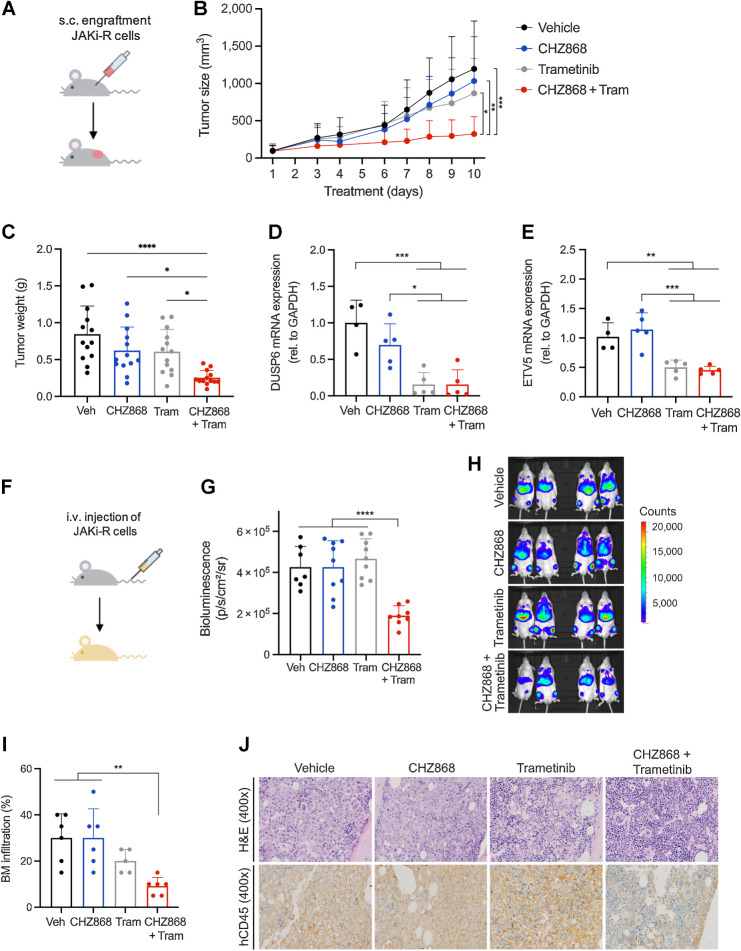
Results: MPN cells showed increased IC50 and reduced apoptosis upon CHZ868 reflecting acquired resistance to JAK2 inhibition. Among >2,500 differential phospho-sites, MAPK pathway activation was most prominent, while JAK2-STAT3/5 remained suppressed. Altered histone occupancy promoting AP-1/GATA binding motif exposure associated with upregulated AXL kinase and enriched RAS target gene profiles. AXL knockdown resensitized MPN cells and combined JAK2/AXL inhibition using bemcentinib or gilteritinib reduced IC50 to levels of sensitive cells. While resistant cells induced tumor growth in NOD/SCID gamma mice despite JAK2 inhibition, JAK2/AXL inhibition largely prevented tumor progression. Because inhibitors of MAPK pathway kinases such as MEK are clinically used in other malignancies, we evaluated JAK2/MAPK inhibition with trametinib to interfere with AXL/MAPK-induced resistance. Tumor growth was halted similarly to JAK2/AXL inhibition and in a systemic cell line-derived mouse model, marrow infiltration was decreased supporting dependency on AXL/MAPK.
Conclusions: We report on a novel mechanism of AXL/MAPK-driven escape from type II JAK2 inhibition, which is targetable at different nodes. This highlights AXL as mediator of acquired resistance warranting inhibition to enhance sustainability of JAK2 inhibition in MPN.
©2023 The Authors; Published by the American Association for Cancer Research.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
