

Reactivation of the G1 enhancer landscape underlies core circuitry addiction to SWI/SNF
- PMID: 37993417
- PMCID: PMC10783513
- DOI: 10.1093/nar/gkad1081
Reactivation of the G1 enhancer landscape underlies core circuitry addiction to SWI/SNF
Abstract
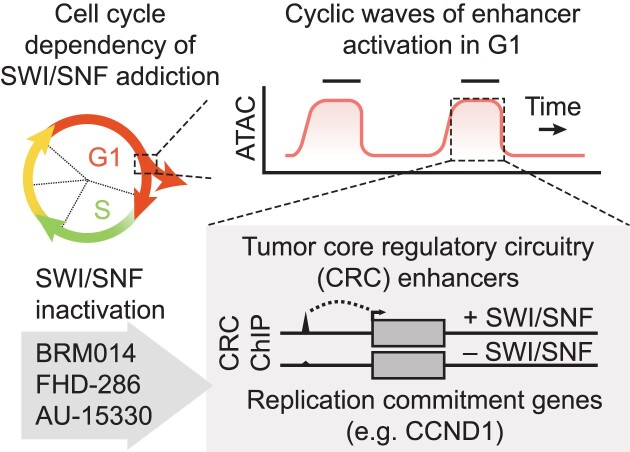
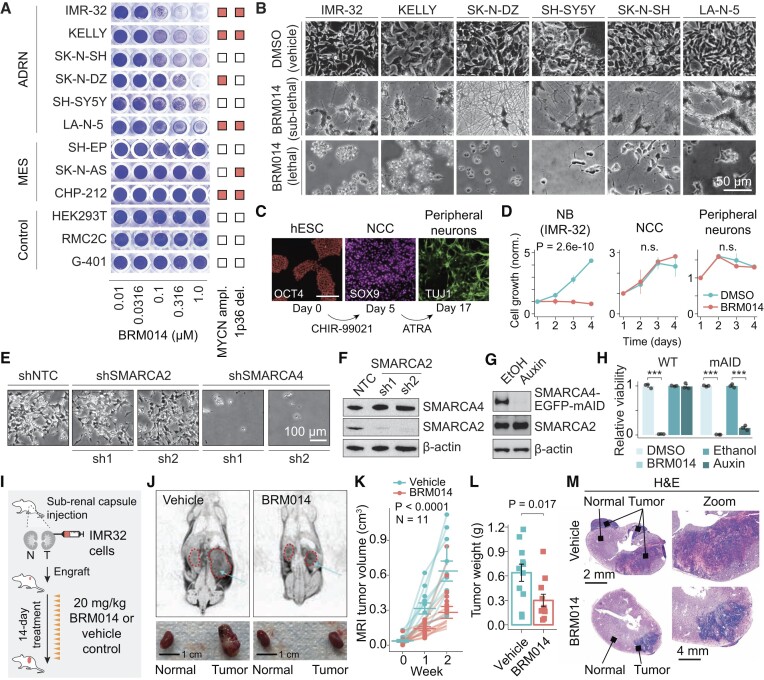
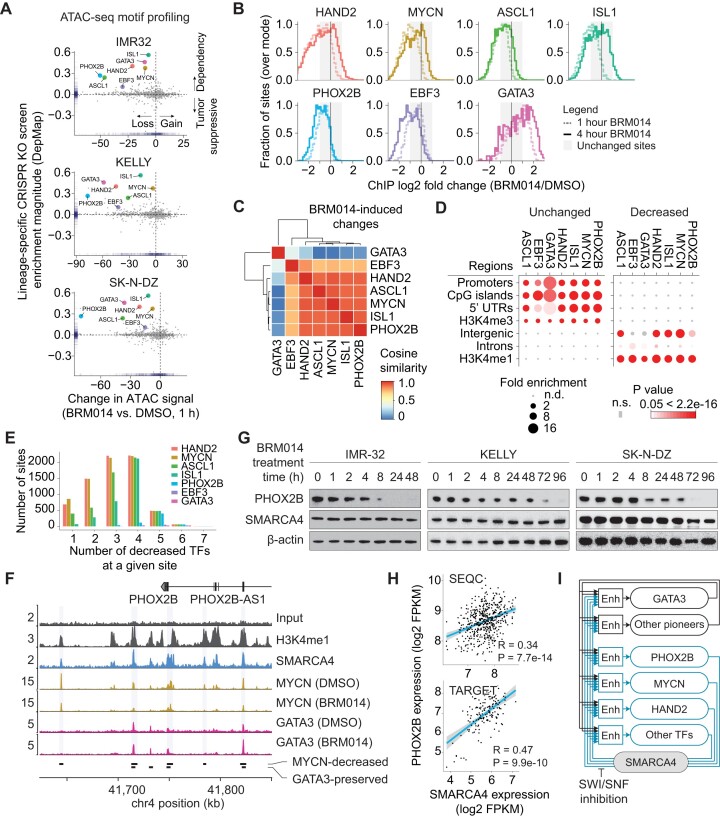
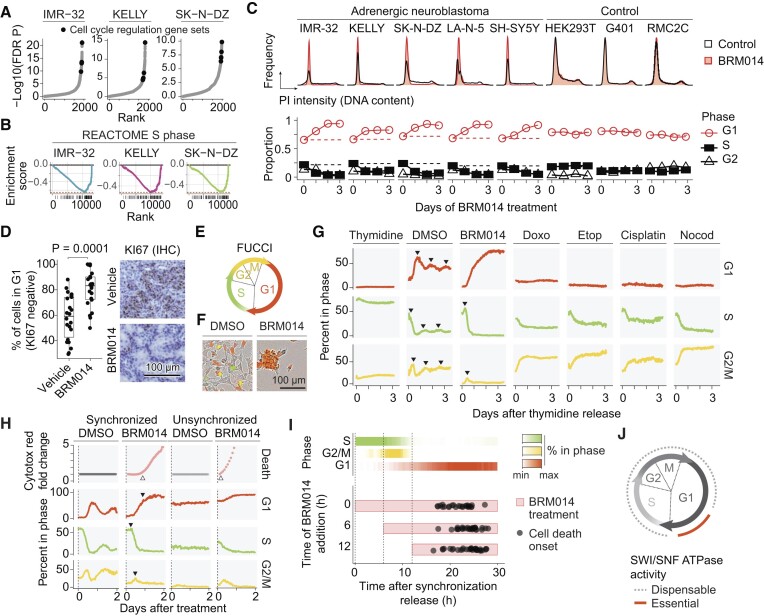
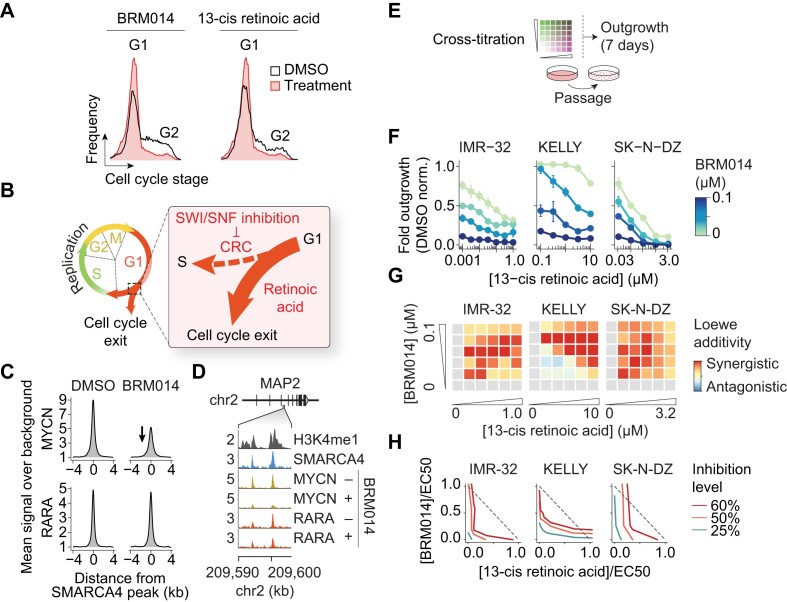
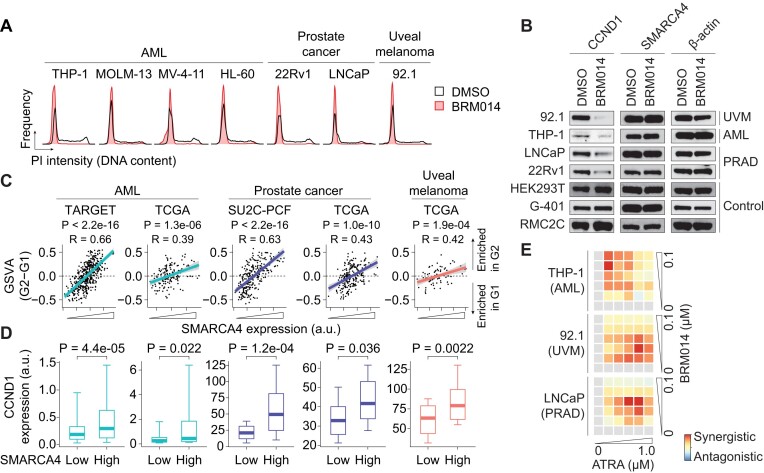
Several cancer core regulatory circuitries (CRCs) depend on the sustained generation of DNA accessibility by SWI/SNF chromatin remodelers. However, the window when SWI/SNF is acutely essential in these settings has not been identified. Here we used neuroblastoma (NB) cells to model and dissect the relationship between cell-cycle progression and SWI/SNF ATPase activity. We find that SWI/SNF inactivation impairs coordinated occupancy of non-pioneer CRC members at enhancers within 1 hour, rapidly breaking their autoregulation. By precisely timing inhibitor treatment following synchronization, we show that SWI/SNF is dispensable for survival in S and G2/M, but becomes acutely essential only during G1 phase. We furthermore developed a new approach to analyze the oscillating patterns of genome-wide DNA accessibility across the cell cycle, which revealed that SWI/SNF-dependent CRC binding sites are enriched at enhancers with peak accessibility during G1 phase, where they activate genes involved in cell-cycle progression. SWI/SNF inhibition strongly impairs G1-S transition and potentiates the ability of retinoids used clinically to induce cell-cycle exit. Similar cell-cycle effects in diverse SWI/SNF-addicted settings highlight G1-S transition as a common cause of SWI/SNF dependency. Our results illustrate that deeper knowledge of the temporal patterns of enhancer-related dependencies may aid the rational targeting of addicted cancers.
Plain language summary
Cancer cells driven by runaway transcription factor networks frequently depend on the cellular machinery that promotes DNA accessibility. For this reason, recently developed small molecules that impair SWI/SNF (or BAF) chromatin remodeling activity have been under active evaluation as anti-cancer agents. However, exactly when SWI/SNF activity is essential in dependent cancers has remained unknown. By combining live-cell imaging and genome-wide profiling in neuroblastoma cells, Cermakova et al. discover that SWI/SNF activity is needed for survival only during G1 phase of the cell cycle. The authors reveal that in several cancer settings, dependency on SWI/SNF arises from the need to reactivate factors involved in G1-S transition. Because of this role, authors find that SWI/SNF inhibition potentiates cell-cycle exit by retinoic acid.
© The Author(s) 2023. Published by Oxford University Press on behalf of Nucleic Acids Research.
Conflict of interest statement
None declared.
Figures

References
-
- Jubierre L., Soriano A., Planells-Ferrer L., París-Coderch L., Tenbaum S.P., Romero O.A., Moubarak R.S., Almazán-Moga A., Molist C., Roma J.et al.. BRG1/SMARCA4 is essential for neuroblastoma cell viability through modulation of cell death and survival pathways. Oncogene. 2016; 35:5179–5190. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
